
Пятна на коже цвета кофе с молоком что это
Обновлено: 28.04.2024
Васкулиты кожи — группа заболеваний мультифакторной природы, при которых ведущим признаком является воспаление кровеносных сосудов дермы и подкожной клетчатки.
Васкулиты кожи — группа заболеваний мультифакторной природы, при которых ведущим признаком является воспаление кровеносных сосудов дермы и подкожной клетчатки.
Трудность в освещении этой темы заключается в том, что до настоящего времени нет общепринятой классификации и даже согласованной терминологии васкулитов. В настоящее время описано около 50 различных нозологических форм, и разобраться в этом многообразии непросто. Пестрота клинических проявлений и недостаточно изученные патогенетические механизмы привели к тому, что под разными названиями может скрываться лишь вариант основного типа поражения кожи. Также, помимо первичных васкулитов, в основе которых лежит воспалительное поражение сосудов кожи, выделяют и вторичные васкулиты (специфические и неспецифические), развивающиеся на фоне определенного инфекционного (сифилис, туберкулез и др.), токсического, паранеопластического или аутоиммунного (системная красная волчанка, дерматомиозит и др.) процесса. Возможна трансформация васкулита кожи в системный процесс с поражением внутренних органов и развитием тяжелых, иногда опасных для жизни осложнений.
Васкулиты кожи — заболевания полиэтиологические. Наиболее часто наблюдается связь с фокальной инфекцией (стрептококки, стафилококки, микобактерии туберкулеза, дрожжевые грибы, вирусы и др.). Определенное значение имеет повышенная чувствительность к ряду лекарственных веществ, в частности к антибиотикам и сульфаниламидным препаратам. Нередко, несмотря на тщательно собранный анамнез и проведенное обследование, этиологический фактор остается невыясненным. Среди факторов риска при васкулитах следует учитывать: возраст (наиболее уязвимы дети и пожилые люди), переохлаждение, чрезмерную инсоляцию, тяжелые физические и психические нагрузки, травмы, операции, заболевания печени, сахарный диабет, гипертонию. Патогенетическим механизмом развития васкулитов кожи в настоящее время считается образование циркулирующих иммунных комплексов с последующей их фиксацией в эндотелии, хотя окончательно это доказано не для всех заболеваний данной группы.
Васкулиты кожи — это неоднородная группа заболеваний, и клинические проявления их чрезвычайно разнообразны. Однако существует целый ряд общих признаков, объединяющих эти дерматозы:
1) воспалительный характер изменений кожи;
2) симметричность высыпаний;
3) склонность к отеку, кровоизлияниям и некрозу;
4) первичная локализация на нижних конечностях;
5) эволюционный полиморфизм;
6) связь с предшествующими инфекционными заболеваниями, приемом лекарств, переохлаждением, аллергическими или аутоиммунными заболеваниями, с нарушением венозного оттока;
7) острое или обостряющееся течение.
Поражения кожи при васкулитах многообразны. Это могут быть пятна, пурпура, узелки, узлы, некрозы, корки, эрозии, язвы и др., но основным клиническим дифференциальным признаком является пальпируемая пурпура (геморрагическая сыпь, возвышающаяся над поверхностью кожи и ощущаемая при пальпации).
Общепринятой классификации васкулитов не существует. Систематизируют васкулиты по разным принципам: этиологии и патогенезу, гистологической картине, остроте процесса, особенностям клинических проявлений. Большинство клиницистов пользуются преимущественно морфологическими классификациями кожных васкулитов, в основу которых обычно положены клинические изменения кожи, а также глубина расположения (и соответственно калибр) пораженных сосудов. Выделяют поверхностные (поражение сосудов дермы) и глубокие (поражение сосудов на границе кожи и подкожной клетчатки) васкулиты. К поверхностным относят: геморрагический васкулит (болезнь Шенлейна–Геноха), аллергический артериолит (полиморфный дермальный ангиит), лейкокластический геморрагический микробид Мишера–Шторка, а также хронические капилляриты (гемосидерозы): кольцевидная телеангиэктатическая пурпура Майокки и болезнь Шамберга. К глубоким: кожную форму узелкового периартериита, острые и хронические узловатые эритемы.
Геморрагический васкулит — системное заболевание, поражающее мелкие сосуды дермы и проявляющееся пальпируемой пурпурой, артралгиями, поражением желудочно-кишечного тракта (ЖКТ) и гломерулонефритом. Встречается в любом возрасте, но наибольшему риску подвергаются мальчики в возрасте от 4 до 8 лет. Развивается после инфекционного заболевания, через 10–20 дней. Острое начало заболевания, с повышением температуры и симптомами интоксикации чаще всего наблюдается в детском возрасте. Выделяют следующие формы геморрагического васкулита: кожная, кожно-суставная, кожно-почечная, абдоминально-кожная и смешанная. Течение может быть молниеносным, острым и затяжным. Длительность заболевания различна — от нескольких недель до нескольких лет.
Процесс начинается симметрично на нижних конечностях и ягодицах. Высыпания имеют папулезно-геморрагический характер, нередко с уртикарными элементами, при надавливании не исчезают. Окраска их меняется в зависимости от времени появления. Высыпания возникают волнообразно (1 раз в 6–8 дней), наиболее бурными бывают первые волны сыпи. Суставной синдром появляется либо одновременно с поражением кожи, либо через несколько часов. Чаще всего поражаются крупные суставы (коленные и голеностопные).
Одним из вариантов заболевания является так называемая некротическая пурпура, наблюдаемая при быстром течении процесса, при котором появляются некротические поражения кожи, изъязвления, геморрагические корки.
Наибольшие трудности вызывает диагностика абдоминальной формы геморрагического васкулита, так как высыпания на коже не всегда предшествуют желудочно-кишечным явлениям (рвоте, схваткообразным болям в животе, напряжению и болезненности его при пальпации, кровью в стуле).
Почечная форма проявляется нарушением деятельности почек различной степени выраженности, от кратковременной нестойкой гематурии и альбуминурии до выраженной картины острого гломерулонефрита. Это поздний симптом, он никогда не встречается до поражения кожи.
Молниеносная форма геморрагического васкулита характеризуется крайне тяжелым течением, высокой лихорадкой, распространенными высыпаниями на коже и слизистых, висцерапатиями, может закончиться смертью больного.
Диагностика заболевания базируется на типичных клинических проявлениях, в атипичных случаях проводится биопсия. При абдоминальной форме необходимо наблюдение хирурга. Рекомендуется наблюдение нефролога в течение трех месяцев после разрешения пурпуры.
Термином «аллергический артериолит» Ruiter (1948) предложил называть несколько родственных форм васкулитов, отличающихся клиническими проявлениями, но имеющих ряд общих этиологических, патогенетических и морфологических признаков.
Патогенетическими факторами болезни считают простуду, фокальные инфекции. Высыпания располагаются обычно симметрично и имеют полиморфный характер (пятна, папулы, пузырьки, пустулы, некрозы, изъязвления, телеангиэктазии, волдыри). В зависимости от преобладающих элементов выделяют три формы заболевания: геморрагический тип, полиморфно-узелковый (соответствует трехсимптомной болезни Гужеро–Дюперра) и узелково?некротический (соответствует узелково?некротическому дерматиту Вертера–Дюмлинга). При регрессе сыпи могут оставаться рубцовые атрофии и рубчики. Заболевание склонно к рецидивам. Нередко перед высыпаниями больные жалуются на недомогание, усталость, головную боль, в разгар заболевания — на боли в суставах (которые иногда припухают) и в животе. Диагностика всех типов заболевания сложна из-за отсутствия типичных, характерных симптомов. При гистологическом исследовании выявляется фибриноидное поражение сосудов мелкого калибра с образованием инфильтративных скоплений из нейтрофилов, эозинофилов, лимфоцитов, плазматических клеток и гистиоцитов.
Геморрагический лейкокластический микробид Мишера–Шторка по клиническому течению сходен с другими формами полиморфных дермальных васкулитов. Признаком, позволяющим выделить это заболевание как самостоятельное, является наличие феномена — лейкоклазии (распад ядер зернистых лейкоцитов, приводящий к образованию ядерной пыли) при гистологическом исследовании. Таким образом, геморрагический лейкокластический микробид может трактоваться как дерматоз, обусловленный хронической фокальной инфекцией (внутрикожные тесты со стрептококковым антигеном положительные), протекающий с выраженной лейкоклазией.
Хронические капилляриты (гемосидерозы), в отличие от остро протекающих пурпур, характеризуются доброкачественным течением и являются исключительно кожными заболеваниями.
Болезнь Шамберга — представляет собой лимфоцитарный капиллярит, который характеризуется наличием петехий и коричневых пурпурных пятен, возникающих чаще всего на нижних конечностях. Пациентов беспокоит исключительно как косметический дефект.
Пурпура Майокки характеризуется появлением на нижних конечностях розовых и ливидно-красных пятен (без предшествующей гиперемии, инфильтрации), медленно растущих с образование кольцевидных фигур. В центральной части пятна развивается небольшая атрофия и ахромия, выпадают пушковые волосы. Субъективные ощущения отсутствуют.
Узелковый периартериит характеризуется некротизирующим воспалением мелких и средних артерий мышечного типа с последующим образованием аневризм сосудов и поражением органов и систем. Наиболее часто встречается у мужчин среднего возраста. Из этиологических факторов важнейшими являются непереносимость лекарств (антибиотиков, сульфаниламидов), вакцинация и персистирование HbsAg в сыворотке крови. Заболевание начинается остро или постепенно с симптомов общего характера — повышения температуры тела, быстро нарастающего похудания, боли в суставах, мышцах, животе, кожных высыпаний, признаков поражения ЖКТ, сердца, периферической нервной системы. Со временем развивается полевисцеральная симптоматика. Особенно характерно для узелкового периартериита поражение почек с развитием гипертонии, которая иногда приобретает злокачественный характер с возникновением почечной недостаточности. Выделяют классическую и кожную форму заболевания. Высыпания на коже представлены узелками — одиночными или группами, плотными, подвижными, болезненными. Характерно образование узлов по ходу артерий, иногда образуют тяжи. Локализация на разгибательных поверхностях голеней и предплечий, на кистях, лице (брови, лоб, углы челюсти) и шеи. Нередко не видны на глаз, могут определяться только пальпаторно. В центре может развиваться некроз с образованием длительно незаживающих язв. Периодически язвы могут кровоточить в течение нескольких часов (симптом «кровоточащего подкожного узла»).
Иногда единственным проявлением заболевания может быть сетчатое или ветвистое ливедо (стойкие фиолетово?красные пятна), локализующиеся на дистальных отделах конечностей, преимущественно на разгибательных поверхностях или пояснице. Характерно обнаружение по ходу ливедо узелков.
Диагностика заболевания основывается на сочетании поражения ряда органов и систем с признаками значительного воспаления, с лихорадкой, изменениями прежде всего в почках, в сердце, наличии полиневрита. Специфических для этой болезни лабораторных показателей не существует. Решающее значение для диагноза имеет динамическое клиническое наблюдение за больным.
Острая узловатая эритема — это панникулит, который характеризуется наличием болезненных розовых узлов на разгибательной поверхности нижних конечностей. Сопровождается лихорадкой, недомоганием, диареей, головной болью, конъюнктивитом и кашлем. Среди взрослых узловатая эритема в 5–6 раз чаще встречается у женщин, пиковый возраст — 20–30 лет. В основе заболевания гиперчуствительность к различным антигенам (бактерии, вирусы, грибы, новообразования и заболевания соединительной ткани). Половина случаев является идиопатическими. Диагностика основывается на данных анамнеза и физического осмотра. Необходимо провести полный анализ крови, рентгенограмму легких (выявляется двусторонняя аденопатия в области корней легких), мазок из зева или быстрый тест на стрептококки.
Хроническая узловая эритема — это группа различных видов узловатых дермогиподермитов. Чаще болеют женщины 30–40 лет. На голенях возникают узлы различной величины с покрасневшей над ними кожей, без наклонности к некрозу и изъязвлению. Воспалительные явления в области высыпаний и субъективные ощущения (артралгии, миалгии) мало выражены. Клинические варианты хронической узловатой эритемы имеют свои особенности, например наклонность узлов к миграции (мигрирующая эритема Беферштедта) или асимметрия процесса (гиподермит Вилановы–Пиноля).
Тактика ведения больного васкулитом кожи
- Классифицировать заболевание (характерная клиническая картина, анамнез, гистологическое исследование).
- Поиск этиологического фактора, но в 30% случаев его установить не удается (поиск очагов хронической инфекции, микробиологические, иммунологические, аллергологические и другие исследования).
- Оценка общего состояния и определение степени активности заболевания: общий анализ крови и мочи, биохимический анализ крови, коагулограмма, иммунограмма. Степень активности васкулитов: I. Высыпания не обильные, температура тела не выше 37,5, общие явления незначительные, СОЭ не выше 25, С-реактивный белок не более ++, комплемент более 30 ед. II. Высыпания обильные (выходят за пределы голени), температура тела выше 37,5, общие явления — головная боль, слабость, симптомы интоксикации, артралгии; СОЭ выше 25, С-реактивный белок более ++, комплемент менее 30 ед., протеинурия.
- Оценка признаков системности (исследование по показаниям).
- Определение вида и режима лечения в зависимости от степени активности: I ст. — возможно лечение в амбулаторных условиях; II ст. — в стационаре. Во всех случаях обострений васкулитов кожи необходим постельный режим, так как у таких больных обычно резко выражен ортостатизм, который следует соблюдать до перехода в регрессирующую стадию. Рекомендуется диета с исключением раздражающей пищи (алкогольные напитки, острые, копченые, соленые и жареные блюда, консервы, шоколад, крепкий чай и кофе, цитрусовые).
- Этиологическое лечение. Если есть возможность устранить причинный агент (лекарство, химикаты, инфекции), то быстро следует разрешение кожных очагов и другого лечения не требуется. Но надо помнить, что при санации очагов инфекции может наблюдаться усиление сосудистого процесса.
- Патогенетическое лечение.
- Профилактические мероприятия: диспансеризация, предупреждение провоцирующих факторов (инфекции, переохлаждение, инсоляции, стрессы и др.), рациональное использование лекарственных средств, трудоустройство, лечебная физкультура, санаторно-курортное лечение.
Лечение геморрагического васкулита
- Глюкокортикостероиды (преднизалон до 1,5 мг/кг) — облегчают проявление кожно-суставного синдрома, но не укорачивают заболевание и не предотвращают поражение почек. Назначаются в тяжелых случаях и под прикрытием гепарина, т. к. повышают свертываемость крови.
- Нестероидные противовоспалительные средства (НПВС) в обычных терапевтических дозировках. Выбор конкретного препарата принципиального значения не имеет (индометацин, диклофенак, ацетилсалициловая кислота).
- Антикоагулянты и антиагреганты. Гепарин при распространенном процессе 300–400 ЕД/кг/сутки. Продолжительность курса должна составлять не менее 3–5 недель. Под контролем коагулограммы.
- Лечебный плазмаферез, когда проявления заболевания не устраняются перечисленными средствами.
- Никотиновая кислота в переносимых дозах в/в капельно.
- Не следует применять: антигистаминные препараты (возможно только в самом начале заболевания), препараты кальция, все витамины.
Лечение васкулитов кожи
1) НПВС (напроксен, диклофенак, Реопирин, индометацин и др.);
2) салицилаты;
3) препараты Са;
4) витамины Р, С, антиоксидантный комплекс;
5) сосудорасширяющие средства (ксантинола никотинат, пентоксифиллин);
6) 2% раствор йодида калия по 1 ст. л. 3 раза в день (узловатая эритема);
7) антикоагулянты и антиагреганты;
8) методы детоксикации в/в капельно;
9) глюкокортикостероиды (ГКС) по 30–35 мг/сутки в течение 8–10 дней;
10) цитостатики;
11) ультравысокочастотная терапия, диатермия, индуктотермия, ультразвук с гидрокортизоном, ультрафиолетовое облучение.
Наружное лечение. При эрозивно-язвенных высыпаниях
1) 1–2% растворы анилиновых красителей;
2) эпителизирующие мази (солкосерил);
3) мази, содержащие глюкокортикоиды, и др.;
4) примочки или мази протеолитическими ферментами (Химопсин, Ируксол);
5) апликации Димексида;
При узлах — сухое тепло.
Лечение не должно заканчиваться с исчезновением клинических проявлений заболевания. Оно продолжается до полной нормализации лабораторных показателей, а в последующие полгода-год больным проводится поддерживающее лечение
Нейрофиброматоз - болезнь Реклингхаузена у детей. Клиника и диагностика
Нейрокутанные синдромы — группа гетерогенных заболеваний с сочетанным поражением кожи и ЦНС. Большинство заболеваний имеет наследственный характер и, вероятно, развивается в результате нарушения дифференциации примитивной эктодермы. К группе нейрокутанных синдромов относятся нейрофиброматоз, туберозный склероз, болезнь Стерджа-Вебера, болезнь Клиппеля-Линдау, атаксия-телеангиэктазия, синдром линейного невуса, гипомеланоз Ито и недержание пигмента.
Нейрофиброматоз (НФ), или болезнь Реклингхаузена, — распространенное заболевание с аутосомно-доминантным типом наследования. Оно характеризуется полиморфизмом клинических проявлений, так как в процесс могут вовлекаться практически все системы и органы, и прогрессирующим течением: характерные признаки заболевания могут присутствовать уже при рождении, однако развитие осложнений может быть отсрочено на несколько десятилетий. НФ возникает в результате нарушения дифференциации клеток нервного гребня и процессов миграции на ранних стадиях эмбриогенеза.
Выделяют две различные формы нейрофиброматоза (НФ). Наиболее распространен нейрофиброматоз типа 1 (НФ-1), встречающийся с частотой 1:4000. Диагноз ставится при наличии любых двух из следующих признаков.
1. Шесть пятен цвета кофе с молоком или более размером более 5 мм до полового созревания и более 15 мм после полового созревания. Пятна цвета кофе с молоком — отличительный признак заболевания, они присутствуют практически у 100 % пациентов с НФ, зачастую уже при рождении, но затем их размер и количество увеличиваются, пигментация становится более выраженной, особенно в первые годы жизни. Пятна могут располагаться на разных участках кожи, преимущественно на туловище и конечностях, пятна на лице менее характерны.
2. Веснушчатость
3. Два узелка Лиша на радужке или более. Узелки Лиша — гамартомы, локализованные на радужку лучше выявляются при исследовании с помощью щелевой лампы. Они определяются более чем у 74 % пациентов с НФ-1, но нехарактерны для НФ-2. Вероятность этого симптома увеличивается с возрастом; узелки Лиша определяются только у 5 % пациентов до 3 лет, у 42 % детей в возрасте 3-4 лет и у 100 % взрослых в возрасте 21 год или старше.
4. Две нейрофибромы или более либо одна плексиформная нейрофиброма. Нейрофибромы в типичных случаях возникают на коже, но могут располагаться вдоль стволов периферических нервов и кровеносных сосудов, а также во внутренних органах, включая ЖКТ. Кожные нейрофибромы выявляются, как правило, в подростковом возрасте или во время беременности, что предполагает влияние гормональных факторов. Они обычно небольшого размера, упругой, эластичной консистенции, с легким багрянистым (фиолетовым) оттенком окружающей кожи.
Плексиформные нейрофибромы обычно выявляются при рождении и возникают в результате диффузного утолщения нервных стволов, которые часто локализуются на лице в орбитальной или височной области. Кожа вокруг плексиформной нейрофибромы может быть гиперпигментирована и выглядит темнее, чем пятна цвета кофе с молоком. Плексиформная нейрофиброма может вызывать чрезмерный рост конечности и деформацию соответствующей кости.
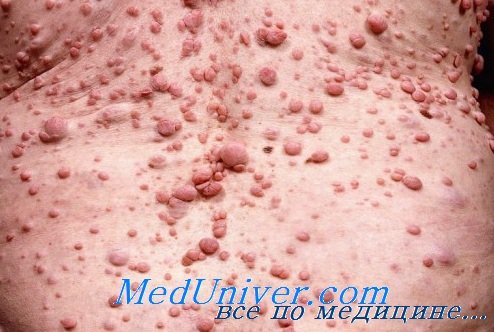
5. Изменения костей. Сколиоз представляет собой одно из наиболее распространенных ортопедических нарушений при НФ-1, хотя это менее специфичный симптом, поэтому он не относится к диагностическим критериям. 6. Глиома зрительного нерва бывает примерно у 15 % пациентов с Нф-I. Эта относительно доброкачественная опухоль состоит из глиозных клеток и муцинозного вещества.
У большинства пациентов с глиомой зрительного нерва клинические проявления отсутствуют; зрение нормальное или почти нормальное, но примерно у 20 % из них выявляются зрительные нарушения или признаки преждевременного полового развития вследствие опухолевой инвазии гипоталамуса. Дети редко ощущают одностороннюю потерю зрения, что может служить причиной поздней диагностики заболевания. У пациентов с односторонней глиомой зрительного нерва как правило нарушена реакция зрачка на свет. Для исследования реакции зрачка на свет каждый глаз поочередно стимулируют ярким светом. На стороне поражения зрачок не сужается, но при освещении здорового глаза возникает равномерное сужение зрачков с двух сторон.
У пациентов с нейрофиброматоза (НФ)-1 и плексиформной невромой века высок риск глиомы зрительного нерва на стороне нейрофибромы. Признаками глиомы зрительного нерва на МРТ служит диффузное утолщение, локальное увеличение или ограниченный объемный процесс, локализованный в области зрительного нерва или хиазмы.
7. У пациента имеются родственники первой степени родства с НФ-1, диагноз которого базируется на приведенных выше критериях. Большинство мутаций при НФ-1 наследуется по отцовской линии. Ген НФ-1 локализован на хромосоме 17qll.2 и кодирует все мРНК размером 11-13 kb, содержащие по крайней мере 59 экзонов, которые продуцируют белок нейрофибрин.
У детей с НФ-1 имеется риск поражения нервной системы. MP-исследование у отдельных детей выявило патологические сигналы в области бледного шара, таламуса и внутренней капсулы, что может быть признаком низкодифференцированной глиомы или гамартомы, не выявляемой на КТ. Эти изменения могут быть ассоциированы с частым снижением школьной успеваемости, синдромом дефицита внимания и речевых нарушений у детей с НФ-1. Сложные парциальные и генерализованные тонико-клонические приступы — частое осложнение заболевания.
Гидроцефалия — редкий симптом, возникающий вследствие стеноза водопровода мозга, макроцефалия с нормальным размером желудочков встречается часто. Возможно поражение сосудов мозга с развитием аневризмы или стеноза вследствие болезни моя-моя. Возможны неврологические нарушения, включая транзиторные ишемические атаки, гемипарез, и когнитивные нарушения. Неудивительно, что выраженность психических нарушений зависит от тяжести заболевания и полиморфизма клинической картины. Признаки преждевременного полового созревания могут быть выражены как в ассоциации с поражением хиазмы зрительных нервов и гипоталамуса, так и при его отсутствии.
Злокачественные опухоли также относятся к тяжелым осложнениям НФ-1. Нейрофиброма в некоторых случаях может перерождаться в нейрофибросаркому или злокачественную шванному. У пациентов с НФ-1 высок риск гипертензии в результате стеноза сосудов почек или феохромоцитомы. Частота развития феохромоцитомы, рабдомиосаркомы, лейкоза, опухоли Вильмса выше у пациентов с НФ-1, чем в общей популяции. Описано необычное сочетание миелоидного лейкоза, ювенильной ксантогранулемы и НФ-1. Опухоли ЦНС, включая глиому зрительных нервов, менингиому ствола и спинного мозга, нейрофиброму, астроцитому и невриному, ответственны за значительную морбидность и летальность в связи с повышением их распространенности у пациентов с НФ-1.
Тип 2 диагностируется примерно у 10 % пациентов с нейрофиброматозом (НФ), встречается с частотой 1:50 000. Диагноз может быть установлен при наличии хотя бы одного из следующих признаков:
1) билатеральные новообразования VIII черепного нерва, представляющие собой невриномы слухового (VIII) нерва, выявляемые на КТ или МРТ;
2) наличие родственников первой степени родства (родителей, детей, сиблингов) с НФ-2 в сочетании с унилатеральным новообразованием VIII черепного нерва или любыми двумя из следующих признаков: нейрофиброма, менингиома, глиома, шваннома или ювенильное заднее подкапсульное помутнение хрусталика.
Билатеральная невринома слухового нерва — основной отличительный симптом НФ-2. Нарушение слуха, слабость мимических мышц, головная боль и нарушение координации могут появиться в детском возрасте, признаки объемного процесса в мостомозжечковом углу чаще выявляются на 2-м или 3-м десятилетии жизни. Хотя пятна цвета кофе с молоком и кожные нейрофибромы служат классическим проявлением НФ-1, они значительно реже встречаются при НФ-2. Заднее подкапсульное помутнение хрусталика выявляется примерно у 50 % пациентов с НФ-2. Как и при НФ-1, опухоли ЦНС, включая шванномы, глиальные опухоли и менингиомы, часто встречаются у пациентов с НФ-2. Ген НФ-2, по данным хромосомного анализа, локализован рядом с центром длинного плеча хромосомы 22ql.ll.
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
БЕСПЛАТНАЯ КОНСУЛЬТАЦИЯ: поможем врачам и владельцам клиник выбрать оборудование для лечения кофейных пятен
Оглавление
Кофейные пятна (пятна цвета кофе с молоком, café au lait spots, café au lait macules, CALMs) — это гиперпигментированные участки на коже с неровными или сглаженными краями, цвет которых варьируется по от светло- до темно-коричневого. В зарубежной литературе их часто называют «café au lait spots», что является сочетанием французского «café au lait» (кофе с молоком) и английского «spots» (пятна).
В нашей компании Вы можете приобрести следующее оборудование для удаления кофейных пятен:
У новорожденных кофейные пятна на теле возникают с различной вероятностью, в зависимости от фототипа кожи:
- 0,3% детей — I–III фототипы;
- 3% детей — IV фототип;
- 18% детей — V–VI фототипы.
До наступления пубертата такие пятна могут появиться с вероятностью 13% у светлокожего населения и 27% — у темнокожего.
Этиология и патогенез
Кофейные пятна на коже обычно говорят о наличии у пациента нейрофиброматоза I типа — мультисистемного заболевания, для которого характерны пятна на спине, шее, в подмышечных областях, а также скелетные дисплазии, доброкачественные или злокачественные опухоли из оболочек нервов (нейрофибромы). Средняя частота появления нейрофиброматоза I типа составляет 1:3000. Его основными причинами считаются мутации или делеции (потери участка) гена NF1. Этот ген несет ответственность за продукцию нейрофибромина 1 — белка, который подавляет возникновение опухолей нервной системы. Снижение его синтеза ведет к появлению нейрофибром и других клинических признаков заболевания.
Кофейные пятна на коже также могут возникать по следующим причинам:
- Синдром Олбрайта-МакКьюна-Штернберга — преждевременное половое созревание, множественная фиброзная остеодисплазия, гиперпигментация кожи.
- Болезнь Бурневилля-Прингла — туберкулезный склероз; редкое генетическое заболевание, при котором в органах и тканях образуются множественные доброкачественные опухоли.
- Анемия Фанкони — проявляется комплексом симптомов, наиболее серьезными из которых являются гематологические нарушения и опухоли.
- Синдром Коффина-Сириса — характеризуется аплазией или гипоплазией дистальной фаланги или ногтя на V пальцах стоп, задержкой в развитии, умственной отсталостью, грубыми чертами лица и др.
Пятна на коже цвета кофе с молоком могут быть следствием RAS-мутаций — изменений в семействе генов и белков RAS. Это малые ГТФазы (ферменты, связывающие и гидролизующие гуанозинтрифосфат — энергетический субстрат для РНК), которые участвуют в передаче сигнала извне клетки и регулируют клеточное деление. Некоторые мутации RAS способствуют появлению и метастазированию опухолей.
У 50% пациентов генетические мутации, ответственные за возникновение кофейных пятен на коже, происходят спонтанно. То есть наследственный механизм передачи заболевания в данном случае имеет опосредованное значение.
Что касается патогенеза, то кофейные пятна появляются на фоне роста продукции меланина и наличия в коже гигантских меланосом. У пациентов с нейрофиброматозом I типа число меланоцитов в пигментных пятнах увеличивается более значительно по сравнению с другими заболеваниями.
Клинические проявления синдрома кофейных пятен
Диагностика нейрофиброматоза I типа строится на поиске ключевых критериев — о заболевании можно говорить при наличии как минимум 2 из 7 признаков:
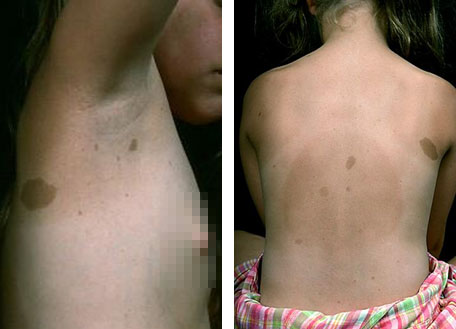
- Шесть и более кофейных пятен или макул на коже диаметром более 5 мм у детей (до пубертата) и более 15 мм у подростков и взрослых (после пубертата) (рис. 1).
- Более двух пятен в паху или в подмышечных областях.
- Две и более типичные нейрофибромы или одна плексиформная нейрофиброма.
- Глиома зрительного нерва.
- Две и более гамартомы радужки глаза — обычно выявляются офтальмологом с помощью щелевой лампы.
- Дисплазия клиновидной кости черепа или патологии длинных костей скелета (например, псевдоартроз).
- Семейный анамнез — наличие нейрофиброматоза у ближайших родственников (отца или матери, братьев или сестер).
Многие из этих признаков отсутствуют до наступления пубертата, что затрудняет диагностику у детей. При этом если пациент из группы риска достигает 10-летнего возраста без появления хотя бы одного симптома (например, кофейных пятен на коже), в дальнейшем он почти наверняка не будет иметь нейрофиброматоза I типа.
Нейрофиброматоз I типа следует отличать от II типа, при котором кожные проявления сравнительно бедные, зато у пациентов имеется склонность к возникновению менингеом и билатеральных акустических неврином.
Другие синдромы, при которых на коже могут возникать кофейные пятна (часть из них была указана выше):
- Синдром Сильвера-Расселла — задержка роста и психомоторного развития, треугольное лицо, укорочение пальцев рук, синдактилия, характерные светло-коричневые пятна на коже и др.
- Синдром Блума — карликовость, сниженный иммунитет, повышенная фоточувствительность кожи, кофейные пятна и др.
- Синдром Горлина-Гольца — наследственная базальноклеточная карцинома кожи с эритематозными поражениями и аномалиями скелета.
- Синдром Маффуччи — доброкачественное разрастание хрящевой ткани с деформацией костей и темными сосудистыми образованиями на коже.
Это далеко не все синдромы, при которых на коже может возникать характерная гиперпигментация цвета кофе с молоком. Однако их объединяет два важных фактора — относительная редкость и общая тяжесть, т.е. наличие других признаков (кроме кофейных пятен), оказывающих гораздо более значимое влияние на организм.
В целом отмечены следующие закономерности:
- Множественные кофейные пятна на теле небольшого размера говорят о наличии одного из вышеназванных заболеваний (обычно это нейрофиброматоз I типа).
- Единичные крупные светло-коричневые пятна, как правило, не связаны с другими патологиями и существуют сами по себе.
Рис. 1. Кофейные пятна в подмышечной области и на спине (www.medscape.com)
Принципы лечения кофейных пятен на теле
До начала терапии кофейных пятен на коже следует исключить более серьезные патологии. Для этого пациента необходимо внимательно опросить, тщательно осмотреть, назначить лабораторные и инструментальные исследования. Сделать это в косметологическом кабинете невозможно, поэтому важно соблюдать преемственность и поддерживать связь с врачами других специальностей.
Одной из таких методик является Q-switched александритовый лазер. В исследовании с участием 48 человек он показал хорошую и отличную эффективность у более половины пациентов (51,4%) после в среднем 3,2 сеансов. При этом рецидив зафиксирован в 10,4% случаев. Лазерное лечение кофейных пятен несет в себе высокий риск поствоспалительной гиперпигментации — около 50%. Если она произошла, следует прекратить терапию до тех пор, пока гиперпигментация не пройдет.
Для лечения кофейных пятен также с успехом применяется IPL-терапия. Интенсивный импульсный свет (IPL) избирательно разрушает меланин в участках его избыточного накопления, что приводит к осветлению кожи. Современные IPL-аппараты, такие как M22 (Lumenis), снабжаются светофильтрами, которые позволяют воздействовать на кожу необходимым диапазоном спектра излучения. Благодаря этому, IPL можно безопасно применять для лечения кофейных пятен на темной коже — вплоть до V фототипа по Фитцпатрику.
Замечено, что если кофейное пятно удалено полностью, то риск его повторного появления минимален. Если же пятно удалено частично, вероятность рецидива составляет 50%.
Пятна цвета кофе с молоком (кофейные пятна)
Синонимы: невус «кофе с молоком», пятна цвета «кофе со сливками», кофейные пятна.
Определение. Гиперпигментированные пятна желто-коричневого цвета, возникающие вследствие гиперпигментации базального слоя эпидермиса.
Этиология и патогенез точно неизвестны.
Частота. В 99,5% случаев кофейные пятна встречаются у обычных людей. Одиночные пятна (не более трех) обнаруживаются в 10—28% случаев у здоровых пациентов. Более 3—5 пятен цвета «кофе с молоком» диагностируются в 0,5% случаев и являются клиническим признаком различных наследственных заболеваний: нейрофиброматоза, туберозного склероза, синдрома Мак-Кьюна—Олбрайта, Сильвера— Рассела и др..
Возраст и пол. Кофейные пятна существуют с рождения или возникают в первые три года жизни. Пол значения не имеет.
Элементы сыпи. Пятно округлое или овальное, размером от 1 до 20 см и более.
Цвет. Равномерная светло- или темно-коричневая окраска.
Локализация любая.
Гистология. Гиперпигментация базального слоя эпидермиса, в ДОФА-положительных меланоцитах выявляются гигантские гранулы (макро-меланосомы).
Диагноз устанавливается клинически.
Течение и прогноз. Если существование пятна является конституциональной особенностью, то оно, как правило, на протяжении всей жизни не меняется. На фоне этого невуса меланома не развивается.
Лечение не проводится. Можно осветлить или убрать с помощью лазера.
а - Пятно цвета «кофе с молоком» в области бедра.
б - Пятно цвета «кофе с молоком» в области плеча.
в - Крупное пятно цвета «кофе с молоком» с неровными краями у пациента 58 лет. Пятна кофе с молоком а, б - Крупное пятно цвета «кофе с молоком» с неровными краями типа «берега Майна» у молодого человека с синдромом Лешке в области левого бедра (а - сзади, б - спереди).
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
Информация на сайте подлежит консультации лечащим врачом и не заменяет очной консультации с ним.
См. подробнее в пользовательском соглашении.
Нейрофиброматоз 1 типа - методы диагностики, лечения по Европейским рекомендациям
а) Синоним. Нейрофиброматоз 1 типа (НФ1) также известен как периферический нейрофиброматоз (НФ) или болезнь фон Реклингхаузена.
б) Эпидемиология. Наиболее распространенная форма нейрофиброматоза (НФ) составляет около 96% случаев с заболеваемостью I на 4000 живорожденных. Гендерное и расовое преобладание не описано.
в) Этиология. Нейрофиброматоз 1 типа (НФ1) наследуется по аутосомно-доминантному типу почти с полной пенетрантностью, но варьирующей экспрессией. Тем не менее, положительный семейный анамнез отмечается почти у 50% пациентов, остальные 50% случаев, как полагают, представляют собой новые мутации. Участвует ген супрессоров опухолей, Ген расположен в перицентрической области на длинном плече 17-й хромосомы и имеет чрезвычайно высокую скорость спонтанной мутации более чем с 200 вариантами, известными на настоящий момент.
Продукт гена, нейрофибромин, представляет собой большой цитоплазматический белок, существующий в нескольких формах в различных тканях; он, как полагают, взаимодействует с внутриклеточными цитоплазматическими микротрубочками.
г) Диагностика и лечение нейрофиброматоза 1 типа. Из-за отсутствия общепринятого генетического теста диагноз основан на клинических проявлениях. Они не всегда присутствуют с рождения, проявляясь в разные периоды жизни.
1. Кожные проявления:
- Пятна цвета «кофе с молоком»: участки ненормальной пигментации кожи от нескольких миллиметров до нескольких сантиметров с четкими границами. Часто они наблюдаются при рождении и почти всегда проявлются до одного года. - С возрастом пятна могут увеличиваться в размерах и количестве, у 80% взрослых с НФ1 имеется, по крайней мере, шесть пятен. Они встречаются на большинстве участков тела, за исключением ладоней, подошв и головы.
- Веснушки: веснушки диаметром несколько миллиметров обычно находятся в субмаммарной, подмышечной или паховой областях. В отличие от пятен цвета «кофе с молоком», эти поражения могут не проявляться до позднего детства или начала пубертатного периода.
2. Глазные проявления:
- Узелки Пиша: пигментированные (желто-коричневые), гамартомы радужки, легко обнаружить с помощью щелевой лампы. Часто отсутствуют в детстве, узелки Лиша появляются с возрастом и имеются примерно у 94% постпубертатных пациентов. Гистопатологически они представлены массой меланоцитов.
- Другие (необычные) глазные проявления: врожденная глаукома, нейрофибромы век и конъюнктивы, утолщение роговицы, конъюнктивы и цилиарного нерва, астроцитомы сетчатки.
3. Опорно-двигательные нарушения:
- Клиновидная дисплазия крыла основной кости: наиболее типичное костное поражение, приводит к пульсирующему экзофтальму.
- Сколиоз: встречается у 10-20% пациентов, часто проявляется в подростковом возрасте.
- Большеберцовой ложный сустав: может быть устойчивым к лечению с необходимостью ампутации в 80% случаев.
- Другие: истончение коркового слоя кости, гиперплазии конечностей, и (редко) рабдомиосаркома.
4. Неврологические проявления:
- Периферические нейрофибромы: множественные кожные, подкожные и периферические поражения, часто распространяются в торакоабдоминальной области или вокруг соска. Гистологически доброкачественные (злокачественные преобразования редки и могут быть представлены в количестве от нескольких до нескольких тысяч). В основном они представляют собой косметическую проблему с возможностью определенной хирургической коррекции по желанию пациента.
- Плексиформные нейрофибромы: часто поражают большое количество периферических нервов или часть симпатической цепочки с потенциальным обезображиванием (гемигипертрофия руки/ноги) или нарушением функции вовлеченных областей. В целом доброкачественные, но злокачественное преобразование происходит (злокачественная оболочки периферических нервов опухоль: MPNST) в 6% случаев. Для бессимптомных поражений предпочтительна тактика выжидания и наблюдения; результаты лечения с 13-цис-ретиноевой кислотой и интерфероном альфа-2а находятся в стадии оценки.
При симптоматическом течении возможно хирургическое уменьшение/удаление опухоли. Тем не менее, риск местного рецидива высок; помимо этого, многие симптоматические поражения порой включают несколько невральных пучков с высоким риском послеоперационного неврологического дефицита. Для MPNST рекомендуется биопсия с последующей возможной ампутацией конечностей, лучевой терапией и химиотерапией (5-летняя выживаемость: 40%).
- Параспинальные нейрофибромы: наиболее распространенные опухоли, поражающие позвоночник у пациентов с нейрофиброматозом 1 типа (НФ1). Параспинальные нейрофибромы, как правило, возникают из спинальных корешков в шейном и поясничном отделах, они могут врасти в позвоночный канал через межпозвонковые отверстия, в результате чего формируются гантелевидные поражения. Хирургическая резекция показана во всех симптоматических случаях и по рентгенологическим критериям (масс-эффект) для шейно-грудной области (риск вторичной миелопатии в связи со сдавлением спинного мозга).
- Глиомы зрительных путей: наиболее распространенное проявление в центральной нервной системе при нейрофиброматозе 1 типа (НФ1), поражает примерно 15% пациентов. Эта опухоль обычно встречается в детском возрасте с наибольшим риском возникновения в течение первых шести лет жизни; женщины страдают вдвое чаще, чем мужчины. Развитие возможно в любой точке зрительного пути, но чаще всего встречаются прехиазмальные поражения. Приблизительно половина всех оптических глиом протекает бессимптомно, и большинство из них, в том числе симптоматические, редко прогрессируют.
Глазные признаки являются наиболее частыми клиническими проявлениями; у 39% детей с поражениями хиазмы выявляется гипоталамо-гипофизарная дисфункция.
д) Рекомендации:
— Бессимптомные случаи: регулярный офтальмологический осмотр, ежегодно в возрасте до шести лет. При выявлении аномалий проводится МРТ мозга и глазниц.
— Симптоматические случаи: обязательно постоянное наблюдение с офтальмологическими осмотрами и МРТ с контрастным усилением каждые три месяца в течение первых 18 месяцев, с последующими контрольными наблюдениями через определенные интервалы при стабильном состоянии.
Хирургическое лечение ограничено определенными показаниями: у детей с внутриглазничным и прехиазмальным поражениями и закрывающимися глазами (или проптоз глаза с тяжелыми формами нарушения зрения) операция может быть выполнена в косметических целях и для предотвращения распространение опухоли на хиазму. При хиазмальных опухолях вмешательство может потребоваться для уменьшения объема больших опухолей, особенно с кистозным компонентом.
У детей старше трех лет лучевая терапия является вариантом лечения, почти 80% случаев дают стабилизацию/уменьшение опухоли. Тем не менее, и в таких случаях при лучевой терапии существует риск нейрокогнигивных и нейроэндокринных последствий, помимо вызываемых облучением злокачественных новообразований. За последние 15 лет ряд клинических исследований показал, что прогрессирование опухоли может быть приостановлено при использовании одно- и мультиагентной химиотерапии, и, следовательно, химиотерапия в настоящее время предлагается в качест ве терапии первой линии, особенно у более молодых пациентов (моложе трех лет), для которых лучевая терапия противопоказана.
- Аномалии ликворных пространств: некоторое расширение желудочков относительно часто выявляется при нейрофиброматозе 1 типа (НФ1), но его роль в патогенезе задержки психомоторного развития спорна. Действительно, в большинстве случаев расширение желудочковой системы не связано с повышением внутричерепного давления и не является прогрессирующим. Это относится и к расширению субарахноидальных влагалищ корешков спинного мозга. Лишь в исключительных случаях эти менингеальные растяжения могут стать симптоматическими при сжатии или деформации корешков спинного мозга, следовательно, будет необходимо лечение (как правило, шунтирование).
- Другие внутричерепные опухоли: глиомы полушарий встречаются менее чем в 0,5%, а глиомы задней ямки менее, чем в 1% случаев нейрофиброматоза 1 типа (НФ1). Большинство этих опухолей не имеет тенденции к росту. По этой причине, как и для поражений зрительных путей, последовательный мониторинг (клинический и рентгенологический) целесообразен в бессимптомных случаях, а оперативное вмешательство показано для пациентов с симптомами и/или опухолями с тенденцией к росту. Неопознанные яркие включения при МРТ — частое явление у больных с НФ1.
Они представлены областями спонтанной гиперинтенсивности на Т2-взвешенных изображениях, чаще всего с участием базальных ганглиев, в мозжечке, в стволе головного мозга и подкорковом белом веществе. Предполагается, что они представляют собой участки миелинопатии, которые, как правило, разрешаются спонтанно.
- Неврологическая задержка развития: 30-65% пациентов с нейрофиброматозом 1 типа (НФ1) проявляют некоторую степень неспособности к обучению, речевые навыки сохраняются лучше, чем визуальнопространственные.
е) Прогноз. При самом лучшем медицинском обслуживании продолжительности жизни при нейрофиброматозе 1 типа (НФ1) остается примерно на 15 лет меньше, чем у населения в целом. Озлокачествление является одной из основных причин снижения ожидаемой продолжительности жизни у взрослых с НФ1, а общий риск злокачественных осложнений составляет примерно 3-15%.
Национальный институт здоровья (NIH),
итоговая конференция по критериям диагностики для нейрофиброматоза (НФ1);
время появления клинических признаков. A-В Сагиттальная (А), аксиальная (Б) и коронарная (В) МРТ в режиме Т1 после введения гадолиния 6-месячному ребенку с нейрофиброматозом 1 типа (НФ1) и диффузной глиомой зрительных путей.
Как это часто случается у такого рода пациентов, рост опухоли вовлек оба зрительных нерва, хиазму и зрительные тракты.
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
Читайте также: