
Склеродермия при гепатите с
Обновлено: 28.04.2024
Кожно-венерологический диспансер №16, Москва
Московский научно-практический центр дерматовенерологии и косметологии Департамента здравоохранения
ГКБ №14 им. В.Г. Короленко Департамента здравоохранения Москвы
Центр теоретических проблем физико-химической фармакологии РАН
Первый МГМУ им. И.М. Сеченова
Склеродермия у женщин
Журнал: Клиническая дерматология и венерология. 2016;15(4): 88‑92
Склеродермия — заболевание соединительной ткани неизвестной этиологии, которое характеризуется фиброзом кожи, поражением сосудов, воспалением, аутоиммунной и тканевой реконструкцией. Мы обследовали 89 пациенток, страдающих склеродермией и проходящих лечение в стационаре филиала «Клиника Короленко» в 2012—2013 гг. Мы обратили внимание на наличие сопутствующих заболеваний у наблюдаемых нами пациенток и сравнили их с описанными в специальной литературе.
Кожно-венерологический диспансер №16, Москва
Московский научно-практический центр дерматовенерологии и косметологии Департамента здравоохранения
ГКБ №14 им. В.Г. Короленко Департамента здравоохранения Москвы
Центр теоретических проблем физико-химической фармакологии РАН
Первый МГМУ им. И.М. Сеченова
Склеродермия является хроническим воспалительным заболеванием соединительной ткани с длительным и прогрессирующим течением, проявляющееся изменениями на коже и внутренних органах. Этиология данного заболевания до конца не изучена. Важное значение в развитии дерматоза играют иммунные и генетические факторы, поражение мелких сосудов, повышенный синтез коллагена фибробластами, а также прием некоторых лекарственных средств, наличие сопутствующих соматических патологий, например, онкологических заболеваний. Склеродермия статистически чаще встречается у женщин в возрасте 20—50 лет.
Мы обследовали 89 пациенток, страдающих склеродермией и госпитализированных для лечения в стационар филиала «Клиника Короленко» в 2012—2013 гг. Мы обратили внимание на наличие сопутствующих заболеваний у наблюдаемых нами пациенток и сравнили их с описанными в специальной литературе.
M. González-López и соавт. [1] проследили связь между системной склеродермией (СС) и первичным билиарным циррозом. В специальной литературе было описано только 6 таких случаев и все они наблюдались у женщин. Авторы описали случай 62-летнего мужчины с множественными воспалительными пятнами на туловище и конечностях, которые появились тремя годами ранее. Клинические и лабораторные данные, а также гистологические исследования соответствовали диагнозу С.С. Сосуществование у мужчины двух болезней, которые преимущественно поражают женщин, подтверждают гипотезу, что имеется патогенетическая связь между этими двумя состояниями. Авторы обращают наше внимание на тот факт, что первичный биллиарный цирроз следует искать у всех больных с С.С. Среди наших пациенток не было больных, страдающих первичным билиарным циррозом.
R. Ludwig и соавт. [3] в своей статье рассматривают хроническую венозную недостаточность (ХВН) как потенциальный триггерный фактор при развитии ограниченной склеродермии (ОС). Авторы, ссылаясь на авторитет других исследователей (Seifarth и соавт.) и собственный опыт, описывают случаи развития ОС в области ХВН. Лечение ХВН у таких больных привело к существенному улучшению клинической картины С в области пораженных вен, но не повлияло на высыпания в других местах. Хроническая венозная недостаточность отмечалась у одной нашей пациентки. Кроме того, у 11 обследованных нами больных встречалось варикозное расширение вен нижних конечностей, которое приводит к развитию ХВН.
В специальной литературе часто отмечается связь между аутоиммунными заболеваниями, такими как склеродермия, дискоидная или системная красная волчанка, витилиго, сахарный диабет (СД). M. Yamamoto и соавт. [4] описали случай 49-летней японки, страдающей дискоидной красной волчанкой (ДКВ) на лице. У нее также были отмечены синдром Рейно, припухлость пальцев, высокие титры антинуклеарных антител, а результаты биопсии показали наличие ограниченного типа С.С. Сочетание С.С. и ДКВ достаточно редки, хотя некоторые похожие случаи уже описывались в Японии. У пациентки также отмечался гепатит С. Среди наших пациенток не было больных, страдающих ДКВ и гепатитом С.
C. Bonifati и соавт. [5] описали случай 21-летнего мужчины, страдающего одновременно ЛС на левой верхней конечности и гомолатеральным сегментообразным витилиго на туловище. Так как эти заболевания появились одновременно, а в процесс была вовлечена одна сторона тела, авторы сделали вывод, что, возможно, связь этих двух заболеваний не случайна.
H. Zeglaoui и соавт. [6] описали случай сосуществования у 15-летней девочки четырех аутоиммунных заболеваний: СД, СС, целиакии и системной красной волчанки. Авторы в своей статье обсуждали этиопатогенез и связь между этими заболеваниями.
Существует большое количество работ, описывающих развитие склеродермии на фоне уже существующих онкологических заболеваний. Среди наших пациенток только у одной отмечался рак щитовидной железы в анамнезе.
A. Scope и соавт. [7] исследовали случаи всех больных склеродермией и раком груди, которые были госпитализированы в течение 1980—2002 гг. Полученные данные сравнивались с литературными данными о больных раком груди и склеродермией. Были суммированы клинические данные 65 женщин со злокачественным новообразованием молочной железы и склеродермией, из них 8 пациенток были госпитализированы, а клинические данные 57 больных описывались в литературе. У 29 (44%) женщин склеродермия развилась перед или одновременно с диагностированием рака груди. Хотя в данной группе больных отмечалась высокая частота СС, все-таки высокая частота ОС имела место в группе, где развитие кожного процесса следовало за диагностикой рака груди. Проведенное исследование подчеркивает связь между склеродермией и раком груди.
K. Kikuchi и соавт. [8] за период с 1994 по 2002 г. наблюдали 71 пациента со склеротическими кожными изменениями; у 66 (93%) из них диагностировали СС, а у 5 (7%) авторы диагностировали псевдосклеродермию, связанную с различными злокачественными новообразованиями. Длительность течения заболевания у этих 5 больных была короче, чем у больных с С.С. Наличие антинуклеарных антител, феномен Рейно или проблемы с пищеводом у больных с псевдосклеродермией проявлялись значительно слабее, чем у пациентов с СС. В сыворотке уровень базального фактора роста фибробластов (bFGF) у 5 больных с псевдосклеродермией был значительно повышен по сравнению с контрольной группой.
Случаи одновременного сосуществования ОС и плоскоклеточного рака были несколько раз описаны в специальной литературе. M. Gréco и соавт. [9] описали множественные очаги плоскоклеточного рака на коже ноги, которые появились у больного с ОС.
G. Androutsopoulos и соавт. [10] описали случай 43-летней нерожавшей гречанки в перименопаузе со склеродермией, у которой появились боли в брюшной полости и кровотечение из влагалища. Женщина перенесла гистерэктомию с двусторонним удалением придатков, полным иссечением сальника, удаление аппендицита и ревизию тазовых лимфатических узлов. Гистопатология показала синхронные первичные раковые образования оболочки матки и левого яичника. Пациентка подверглась послеоперационной химиотерапии и оставалась в хорошем состоянии без проявлений болезни спустя 25 мес после операции. Синхронные первичные раковые образования оболочки матки и яичника относительно редки в общей популяции населения. В специальной литературе сообщалось всего о нескольких случаях рака женского полового тракта у пациенток, страдающих склеродермией.
S. Nunez и соавт. [11] сообщили о пациенте со склеродермией, почечно-клеточным раком (ПКР) и мембранозной нефропaтией. Определенные клинические и лабораторные данные позволили предположить, что ПКР вызвал или облегчил развитие двух других состояний. Карцинома верхушки левой почки была удалена при помощи нефрэктомии. Мембранозная нефропaтия была найдена в почечной паренхиме рядом с удаленной опухолью. Через 3,5 года после хирургического вмешательства клинические проявления склеродермии приостановились, хотя количество лекарственных препаратов было сильно уменьшено. Данный случай служит примером развития трех редких заболеваний (склеродермии, ПКР и мембранозной нефропaтии) у одного человека, длительное улучшение проявлений склеродермии и мембранозной нефропaтии, наступившее после удаления ПКР, противопоставленное быстрому ухудшению этих состояний до операции. У пациента в сыворотке присутствовали аутоантитела, которые редко встречаются у больных со склеродермией, что, вероятно, связано со злокачественной опухолью и позволяет предположить патогенетическую связь между этими тремя состояниями.
A. Dancey и соавт. [12] проследили связь между радиоизлучением и развитием склеродермии. Впервые случаи развития склеродермии, вызванные радиоизлучением, были описаны в 1900 г. Случаи склеродермии, вызванные радиацией, встречаются с частотой 2:1000. Распространение процесса за пределы облученного участка наблюдается приблизительно в ¼ случаев. Авторы описали 2 случая склеродермии на коже молочной железы. У первой пациентки развитие склеродермии не было связано с патологией молочной железы. Больной провели операцию по Wise с удалением очага склеродермии в пределах здоровой кожи. У второй больной склеродермия развилась после курса радиотерапии по поводу рака молочной железы. Больная отказалась от хирургического вмешательства. В статье подчеркивается, что склеродермия является признанным осложнением радиотерапии, которое необходимо отличать от склеротического рецидива первичной опухоли.
S. Dubner и соавт. [13] описали случай 52-летней женщины, которой удалили опухоль молочной железы с последующей радиотерапией. Через 2 года после операции на коже в области облучения появились изменения в виде гиперпигментации, втягивания и уплотнения кожи. Клиническая и гистологическая картина позволили диагностировать ОС после облучения. Авторы подчеркнули, что важно помнить об этих редких осложнениях после радиотерапии.
K. Nakamura и соавт. [14] описали случай 65-летней женщины с хронической болезнью «трансплантат против хозяина», проявившейся склеротическими изменениями на коже верхних конечностей и туловища после трансплантации аутологических стволовых клеток переферической крови. Больная страдала раком груди. После хирургического вмешательства и химиотерапии у нее развилась панцитопения, по поводу которой была проведена трансплантация аутологических стволовых клеток периферической крови. Через 4 года после трансплантации у больной развился тяжелый склероз на коже верхних конечностей и туловища. Биопсия кожи показала истончение эпидермиса и пролиферацию коллагеновых пучков в дерме. Отмечалось отсутствие титров антинуклеарных ДНК в сыворотке. Больной был поставлен диагноз: хроническая болезнь «трансплантат против хозяина». Несмотря на лечение преднизолоном per os, кожные склеротические изменения продолжали развиваться и рак груди рецидивировал.
M. Juarez и соавт. [15] описали случай пациента со вторичной склеродермией как проявлением паранеопластического синдрома при волосато-клеточном лейкозе. Также у одной нашей больной в анамнезе имелся волосато-клеточный лейкоз.
Аутоиммунные болезни могут проявляться как паранеопластический синдром при онкологических заболеваниях, чаще всего как дерматомиозит/полимиозит. В данной статье G. Orphanos и соавт. [16] описали случай 56-летнего мужчины с раком прямой кишки и С.С. Развитие этих двух болезней было настолько связано, что склеродермию сочли паранеопластическим синдромом на фоне онкологического заболевания, а не сопутствующим заболеванием.
Среди форм заболевания, наблюдаемых нами у пациенток стационара Городской клинической больницы № 14 им. В.Г. Короленко, отмечались очаговая и линейная склеродермия, склероатрофический лихен, атрофодермия Пазини—Пьерини и синдром Тибьержа—Вейссенбаха.
Клиническая картина ОС представлена единичными или множественными пятнистыми элементами розово-сиреневого цвета с четкой границей округлой или овальной формы. С течением времени цвет элемента меняется с розово-сиреневого на желтовато-белый («цвет слоновой кости»), очаг уплотняется, а позже в нем развивается атрофия. По периферии очага отмечается розово-сиреневый ободок, который свидетельствует об активности процесса. В месте атрофии кожный рисунок сглаживается, чувствительность ослабевает, сало- и потоотделение отсутствуют. Очаг начинает блестеть и постепенно цвет очага меняется на коричневый. Кожа в очаге истончается. Высыпания развиваются на любом участке кожного покрова. Среди наших больных ОС отмечалась у 74 больных.
Линейная склеродермия чаще развивается у молодых женщин или детей. Высыпания чаще всего локализуются на коже головы или конечностей. Свое название данная форма дерматоза получила за форму высыпного элемента, который имеет линейную форму и напоминает рубец после удара сабли. У наблюдаемых нами пациенток очаги линейной склеродермии присутствовали на коже у 5 пациенток.
Клиническая картина склероатрофического лихена представлена мелкими пятнистыми высыпными элементами беловатого цвета, локализованными чаще всего на коже шеи и груди, а также в области половых органов. По периферии пятен может присутствовать венчик розово-сиреневого цвета. С течением времени отмечается западение центральной части пятен и развитие атрофии. Расширенные устья волосяных фолликулов и потовых желез могут быть заполнены роговыми пробками. Также для склероатрофического лихена характерно наличие петехий и телеангиэктазий. На гениталиях склероатрофический лихен представлен очагами сухости, истончения и атрофии кожи, вплоть до сужения входа во влагалище. Пациенток иногда беспокоит зуд. Заболевание чаще развивается у женщин в возрасте 50—60 лет, что, вероятно, связано с гормональными нарушениями и наличием большого количества сопутствующих патологий. Среди наблюдаемых нами пациенток у 8 присутствовали высыпания склероатрофического лихена.
Атрофодермия Пазини—Пьерини характеризуется несколькими пятнами розовато-синюшного цвета округлой или овальной формы. Данным заболеванием чаще страдают молодые женщины. С течением времени свойственное для склеродермии уплотнение не развивается. Очаг приобретает буровато-коричневый оттенок и отмечается атрофия. Венчик по периферии очага, сопутствующий склеродермии, также отсутствует. В связи с отсутствием этих двух симптомов, всегда сопутствующих склеродермии, по мнению некоторых авторов, данное заболевание может служить проявлением поздней стадии иксодового клещевого боррелиоза (М.А. Пальцев и соавт., 2004) [17]. Среди наших пациенток была одна больная с данным диагнозом. В то же время встречаются клинические случаи, когда у одного больного одновременно существуют очаги атрофодермии Пазини—Пьерини и ОС.
Синдром Тибьержа—Вейссенбаха является редким. Он характеризуется сочетанием склеродермии и кальциноза кожи. Среди наших больных только у одной отмечался синдромом Тибьержа—Вейссенбаха.
Среди 89 больных стационара филиала «Клиника Короленко» были женщины в возрасте от 23 до 80 лет. Распределение возраста пациенток представлено на рис. 1. В обследованной группе были 3 пациентки в возрасте от 20 до 30 лет, 7 больных — от 31 года до 40 лет, 6 — 41—50 лет, 23 — 51—61 год, 27 — от 61 года до 70 лет, 23 в возрастном промежутке 71—80 лет.

Рис. 1. Возраст пациенток.
Среди больных у 74 пациенток отмечалась ОС, 8 пациенток страдали склероатрофическим лихеном, 5 больным был выставлен диагноз линейная склеродермия. Одна больная страдала синдромом Тибьержа—Вейссенбаха и у одной пациентки была выявлена атрофодермия Пазини—Пьерини. Распределение форм заболевания представлено на рис. 2.

Рис. 2. Формы заболевания.
Длительность заболевания составляла от 1 мес до 35 лет. С длительностью заболевания до 1 года наблюдались 3 пациентки. У 63 больных длительность заболевания варьировала от 1 года до 5 лет, у 13 пациенток — от 6 до 10 лет, 7 больных — от 11 до 20 лет, 3 пациентки — более 20 лет, из них 1 считала себя больной 26 лет, 1 пациентка — 30 лет и 1 — 35 лет. Распределение длительности заболевания представлено на рис. 3.

Рис. 3. Длительность заболевания.
Сопутствующие патологические процессы отмечались у 89 (100%) человек: заболевания сердечно-сосудистой системы были обнаружены у 45 (50,6%), из них артериальной гипертензией страдали 45 (50,6%), 32 (35,9%) — ишемической болезнью сердца, 6 (6,7%) — нарушениями сердечного ритма. Со стороны желудочно-кишечного тракта (ЖКТ) жалобы поступали от 31 (34,8%) пациентки: у 2 (2,2%) — желчнокаменная болезнь, у 15 (16,9%) — холецистит, у 6 (6,7%) — жировой гепатоз, язвой желудка и двенадцатиперстной кишки страдали по одной (1,1%) пациентке, хронический гастрит отмечался у 30 (33,7%) больных, неспецифическим язвенным колитом страдала 1 (1,1%) пациентка. Заболеваниями почек страдали 11 (12,3%) пациенток: пиелонефритом — 8 (8,9%) пациенток, кистами почек — 5 (5,6%). Заболевания щитовидной железы отмечали у 31 (34,8%) больной: узловой зоб — у 17 (19,1%), гипотиреоз —у 10 (11,2%), аутоиммунный тиреоидит — у 8 (8,9%), рак щитовидной железы имелся в анамнезе у 1 (1,1%) пациентки. СД 2-го типа страдали 17 (19,1%) пациенток, при этом у 11 (12,3%) женщин отмечались осложнения СД, такие как диабетическая ретинопатия у 3 (3,3%), ангиопатия сетчатки у 8 (8,9%) пациенток и диабетическая полинейропатия у 2 (2,2%). Заболеваниями дыхательных путей страдали 10 (11,2%): хроническим бронхитом — 10 (11,2%) пациенток, бронхиальной астмой — 1 (1,1%) и хроническим тонзиллитом — 4 (4,5%). Волосато-клеточный лейкоз отмечался в анамнезе у 1 (1,1%) больной. Сопутствующие заболевания, наблюдаемые нами у обследованных больных, представлены на рис. 4.

Рис. 4. Сопутствующие соматические заболевания.
Патогенез АГ сложен. Полагают, что это ответ генетически предрасположенного организма на какой-то внешний агент, который является пусковым моментом в развитии аутоиммунных процессов, вызывающих прогрессирующие воспалительно-некротические изменени
 |
Патогенез АГ сложен. Полагают, что это ответ генетически предрасположенного организма на какой-то внешний агент, который является пусковым моментом в развитии аутоиммунных процессов, вызывающих прогрессирующие воспалительно-некротические изменения, приводящие к фиброзу и циррозу печени (ЦП). Генетически детерминированная предрасположенность к этому заболеванию выявлена во многих исследованиях. Доказано, что большая часть больных АГ имеют фенотип по антигенам главного комплекса гистосовместимости HLA-B8, HLA-DR4, DR3 и DR52a. Пусковой агент пока неизвестен, однако есть некоторые данные о роли вирусов гепатита [31, 26], кори [27], Эпштейн-Барр вируса [32], а также интерферона (ИФН) [14] как инициаторов начала АГ.
| Аутоиммунный гепатит (АГ) — хроническое воспалительное заболевание печени невыясненой этиологии, характеризующееся определенными лабораторными, клиническими и гистологическими признаками. Болеют им в основном женщины молодого возраста |
АГ — это прогрессирующее воспаление печени, характеризующееся наличием некрозов в перипортальной, септальной зонах (ступенчатые некрозы) или, более широко, лобулярным гепатитом (ЛГ), гипергаммаглобулинемией и аутоантителами в сыворотке крови [7]. Портальные тракты печени на биоптатах находят расширенными с накоплением в них обширных инфильтратов, имеющих разный клеточный состав: лимфоциты, макрофаги, плазматические клетки. ЛГ — дольковый гепатит, когда некрозы выявляются во второй и третьей зонах ацинусов, а также обнаруживается внутридольковая лимфоидноклеточная инфильтрация, которая выражена значительно больше, чем инфильтрация портальных трактов. ЛГ является частью гистологической картины АГ, если он выявляется одновременно с перипортальным гепатитом. По гистологической картине на АГ может указывать, кроме вышеперечисленного, наличие многоядерных гепатоцитов [2].
Наконец, картина фиброза может присутствовать в той или иной степени даже при умеренной степени активности АГ, а в запущенных случаях, особенно при отсутствии эффективной терапии, формируются мостовидные некрозы и, в конце концов, ЦП.
Хотя гистологическая картина при АГ очень характерна, все-таки она неспецифична. Отличительной чертой АГ является обнаружение в биоптатах преимущественно плазматических клеток, так как выраженная инфильтрация в портальной, перипортальной зоне, вовлечение в процесс долек печени — в равной мере присущи и хроническому вирусному гепатиту (ХВГ).
Одной из основных клинических характеристик АГ является обнаружение аутоантител к клеточным и субклеточным структурам клеток разных органов [22]. Типичным маркером АГ являются антитела к ядрам клеток — ANA. Из других маркеров выявляются антитела к клеткам гладкой мускулатуры (SMA), антитела к микросомам клеток печени и эпителиальных клеток клубочкового аппарата почек (LKM), антитела к растворимому печеночному антигену (SLA), антитела к антигенам (цитокератины 8, 18) мембран гепатоцитов — LMA.
Клинические проявления АГ очень разнообразны [1, 3, 4]. С одной стороны, встречаются бессимптомные формы, когда случайно выявляется повышение АЛТ, АСТ, а с другой — острое начало болезни с тяжелым течением вплоть до развития фульминантного гепатита (ФГ).
Нередко заболевание начинается незаметно с астеновегетативных проявлений, болей в области правого предреберья, незначительной желтухи. Однако у большинства больных АГ начало болезни острое, как при остром вирусном гепатите (ОВГ), и при осмотре пациента врач впервые выявляет признаки хронического гепатита (ХГ) — телеангиоэктазии, пальмарную эритему, увеличение печени и селезенки, а также изменения в анализах крови — гипергаммаглобулинемию, увеличение IgG, снижение содержания общего белка, резкое увеличение СОЭ. Лейкопения и тромбоцитопения наблюдаются у больных на поздних стадиях болезни или при развившихся гиперспленизме и синдроме портальной гипертензии.
Когда АГ впервые проявляется желтухой, как при ОВГ, приходится дифференцировать его от гепатитов А, В, Е и особенно С, при котором антитела в сыворотке крови могут появляться через достаточно продолжительное время после начала болезни. Желтуха у пациентов с АГ может быть разной степени выраженности, часто появляется на поздних стадиях заболевания, бывает непостоянной и усиливается в период обострений. В общем же у большинства больных чаще всего изменяются аминотрансферазы, нежели щелочная фосфотаза (ЩФ) или билирубин.
| Аутоиммунный гепатит был выделен из группы болезней печени и впервые описан как отдельная нозология в начале 50-х годов [33]. В научной литературе существовал под разными названиями. Термин люпоидный гепатит, который часто использовался в нашей стране, ввел в 1956 году Дж. Маккей с соавторами в журнале Lancet, так как при этом заболевании нередко в сыворотке крови больных выявлялись волчаночные клетки. Потом, в последующие годы, люпоидный, или классический, АГ стали называть аутоиммунным активным хроническим гепатитом, но в 1993 году Международная группа по изучению болезней печени предложила термин АГ, а также критерии установления его диагноза [17] |
Для АГ характерно поражение кожи в виде геморрагической сыпи, оставляющей после себя пигментацию. Из других симптомов встречаются волчаночная и узловатая эритемы, очаговая склеродермия, пальмарная эритема и телеангиоэктазии. У всех больных выявляются изменения в эндокринной системе — аменорея, угри, гирсутизм, стрии. Диагностическое значение отдельных симптомов болезни при АГ неодинаково. К наиболее значимым относятся длительная лихорадка и арталгии. В большинстве случаев АГ они присутствуют одновременно, являясь наиболее частыми и постоянно встречающимися жалобами больных [4].
Один из вариантов начала АГ — появление лихорадки с внепеченочными проявлениями, из которых следует назвать аутоиммунный тиреоидит, язвенный колит, гипертиреоидизм, гемолитическая анемия, идеопатическая тромбоцитопения, сахарный диабет, целиакия, полимиозит, фиброзирующий альвеолит, гломерулонефрит и т. д. Желтуха при этом варианте появляется позже [20].
Часто АГ сопровождается бесплодием, однако при возникновении беременности и последующих родах на фоне компенсированного процесса это не влияет на течение АГ и судьбу ребенка даже при постоянном приеме преднизолона (ПР) [30]. Беременность на стадии сформировавшегося ЦП и синдрома портальной гипертензии, которые выявляются у трети больных на момент выявления АГ, нежелательна [3].
В отличие от ХВГ течение АГ у больных непрерывно прогрессирующее, без самопроизвольных ремиссий. Улучшения самочувствия бывают кратковременными, нормализации биохимических процессов не происходит. Прогноз течения АГ хуже у пациентов с острым началом болезни по типу ОВГ, с наличием признаков холестаза, асцитом, повторными эпизодами острой печеночной энцефалопатии (ОПЭ). Как правило, больные, пережившие критический период, имеют лучший прогноз.
Диагноз АГ выставляется на основании соответствия лабораторных и гистологических данных, отсутствия маркеров ВГ, исключения злоупотребления алкоголем и контактов с препаратами крови, гипотоксическими веществами, повышения гамма-глобулинов не менее чем в 1,5 раза выше нормы. Повреждение желчных протоков, отложение меди, гемосидероз, при которых также могут выявляться ЛГ и ступенчатые некрозы, предполагают другую причину ХГ и исключают диагноз АГ. ANA, SMA и LRM-1 должны быть в титрах не менее 1:80 у взрослых и 1:20 у детей (рекомендации Международной группы, 1993).
Дифференциальный диагноз между АГ и другими аутоиммунными заболеваниями, в основном первичным билиарным циррозом (ПБЦ), первичным склерозирующим холангитом (ПСХ), ХВГ основывается на клинических, гистологических и иммунологических параметрах. Однако нередко выявляется так называемый overlap-синдром, когда одновременно у пациентов выявляются признаки АГ и вышеперечисленных хронических заболеваний печени. Далее они будут описываться как варианты АГ [7, 13]. Предполагаемый диагноз АГ в данном случае подразумевает сходство с клиникой АГ (жалобы на слабость, арталгии, миалгии), а биохимический анализ крови отражает преимущественно изменения холестатического порядка, имеет место кожный зуд разной степени выраженности. Пациенты с такими вариантами АГ могут быть обоего пола, любого возраста, но все же чаще это женщины в возрасте до 40 лет и моложе. На гистологии находят перипортальный гепатит с или без ЛГ, часто с поражением желчных протоков, жировой дистрофией гепатоцитов и лимфоидной инфильтацией портальных трактов в виде гранулем [7, 10].
Деление АГ на подтипы практического значения не имеет, однако следует иметь в виду, что подтип 2 АГ может быть связан с гепатитом С либо HCV может индуцировать появление АГ у генетически предрасположенных лиц. Нет данных о различиях гистологической картины при отдельных подтипах АГ
Большинство больных с ПБЦ можно точно отделить от пациентов с АГ с помощью характерных лабораторных и иммунологических данных. Однако при этом варианте наряду с характерными параметрами АГ нередко выявляются гистологические признаки холангита и АМА (антитела к антигенам внутренней поверхности мембраны митохондрий), что очень характерно для ПБЦ. Наиболее важным для подтверждения диагноза ПБЦ является обнаружение АМА подтипа М2 [6]. АМА выявляются у 20-27% больных АГ в разных титрах [19]. Это может отражать диагностические ошибки в определении иммуносерологических маркеров, другие заболевания или одну из стадий ПБЦ. Если у больного повышена щелочная фосфатаза (ЩФ), IgM сыворотки крови и обнаружена АМА — вероятен диагноз ПБЦ. Трех-шестимесячный курс лечения стероидами помогает расшифровать преобладающую патологию — при реакции на лечение можно говорить о превалировании АГ.
Установлено, что у 16% больных АГ выявляется язвенный колит (ЯК), наличие которого характерно для пациентов с ПСХ (от 40 до 60% больных). К тому же при таком сочетании — АГ и признаки ПСХ (наличие ЯК, поражение желчных протоков, слабый ответ на стероиды) — также обнаруживают фенотип HLA-B8, HLADR3, HLA DR4. Поэтому наличие кожного зуда у больных АГ и повышение ЩФ более чем в четыре раза против нормы указывают на необходимость проведения холангиографии (ХГР) и вероятность развития варианта АГ и ПСХ. Поражения желчных протоков несовместимы с диагнозом АГ. Они редки, но когда появляются у больных АГ с сопутствующей патологией кишечника или атипичным повышением ЩФ, можно допустить этот вариант АГ. Окончательный диагноз зависит от результатов ХГР. ХГР выявляет признаки склерозирующего холагнита у 42% больных АГ и ЯК. Но иногда ХГР бывает в норме у 14% больных ПСХ при гистологически подтвержденном диагнозе. Об этом необходимо помнить [24].
АГ считается заболеванием невирусной этиологии, но у 4% больных АГ выявляются антиHCV и еще у 4% — маркеры вируса гепатита В. Больные АГ, имеющие атипичное течение болезни либо плохо отвечающие на терапию стероидами, нередко имеют в сыворотке крови HCV RNA. Любопытно, что 11% больных ХВГ имеют SMA и 28% — ANA. У 62% выявляются аутоантитела к щитовидной железе и ревматоидный фактор. Большая часть этих больных имеют низкие титры SMA и ANA (1:80 и ниже), а пациенты с точным диагнозом АГ — SMA в титрах 1:160 и ANA 1:320. Поэтому больные АГ и с выявляемыми SMA или ANA в титрах ниже 1:320 могут быть отнесены к группе с превалированием вирусного заболевания [11].
Тем не менее пациенты с АГ имеют более выраженную инфильтрацию портальных трактов плазматическими клетками, более выраженные воспалительные изменения в дольках и больше ступенчатых и перисептальных некрозов по сравнению с пациентами ХВГ, особенно ХГС. У больных ХВГ/ХГС наоборот — в портальных трактах преобладает лимфоидноклеточная инфильтрация, чаще выявляется стеатоз и повреждения желчных протоков, особенно при ХГС.
У 13% взрослых больных с признаками АГ не обнаруживаются аутоантитела, а все остальные признаки — иммунологические, биохимические и гистологические, а также возраст и пол соответствуют критериям постановки диагноза АГ. Что важно, эти больные также хорошо реагируют на лечение стероидами [8, 9]. Отмечено, что с течением времени при динамическом наблюдении у некоторых из них появляются соответствующие аутоантитела, характерные для АГ.
Несмотря на разнообразие клинической картины, при АГ основой лечения является назначение преднизолона (ПР). Ответ на данную терапию — один из критериев постановки диагноза АГ. Целесообразность назначения ПР при АГ доказана в многочисленных исследованиях и обусловлена редкими самопроизвольными ремиссиями в течении болезни, высокой смертностью и ухудшением качества жизни [12, 18, 23, 28, 29]. При назначении ПР смертность удается снизить в течение пяти лет с 50 до 20%, а частоту индуцированных ремиссий довести до 80%. У большинства больных ремиссии появляются в течение первых двух лет терапии и почти у всех в последующие четыре года лечения.
Лечение ПР следует назначать всем больным АГ высокой степени активности с фиброзом и циррозом или без. У больных с умеренной степенью активности болезни назначение ПР часто определяется наличием жалоб и симптомов болезни. Больные без симптомов и с умеренной степенью активности процесса по гистологической картине не нуждаются в лечении, но должны тщательно и регулярно наблюдаться для своевременного выявления признаков прогрессирования болезни.
Как правило, начальная доза ПР составляет 20-30 мг/сутки с последующим постепенным снижением ее до поддерживающей — обычно 10 мг/сутки. Из всех схем лечения предпочтителен ежедневный прием однократно утром. Осложнения терапии наблюдаются при дозе более 10 мг/сутки. Нет точных рекомендаций по отмене или снижению дозы иммуносупрессоров, некоторые больные могут долго оставаться в ремиссии после отмены ПР.
Однако было установлено, что у большей части больных в дальнейшем, даже спустя несколько лет после ремиссии, появляются признаки обострения и часто требуется большая доза для ее достижения [15].
Комбинация ПР с азатиоприном (АЗА) может уменьшить побочные эффекты (при этом требуется небольшая доза ПР). Лучше давать 10 мг/сутки ПР с 50 мг/сутки АЗА, чем один ПР, но в большей дозе. Сам АЗА не способен индуцировать ремиссию, но его добавление к ПР поддерживает ее даже в дозе 1 мг/кг/сутки. При неэффективности лечения АЗА назначали 6-меркаптопурин с хорошим эффектом [25]. У 20% больных АГ не удается достигнуть ремиссии — чаще всего у пациентов с признаками ЦП, лиц молодого возраста, при длительном анамнезе болезни до начала терапии ПР и у больных с фенотипом HLA-B8, DR3 [28]. Побочные эффекты при назначении иммунодепрессантов редкие, это в основном диспепсический синдром, сыпи, кушингоидизм, нарушение роста и развития у детей, сахарный диабет и остеопороз у женщин в менопаузе. АЗА может индуцировать миелосупрессию, возникновение катаракты, обладает онкогенным и, возможно, тератогенным эффектами.
Лечение вариантов АГ представляет определенные трудности. Основа терапии, препарат выбора для начала лечения — и здесь ПР. При сочетании АГ и ПБЦ назначают ПР в дозе 20 мг/сутки от трех до шести месяцев, а при отсутствии эффекта — урсодезоксихолевую кислоту (УДХК) или ее коммерческие препараты (урсофальк, урсосан, урсодиол и др.) по 13-15 мг/сутки от трех до шести месяцев.
Тактика лечения больных с вариантом АГ и ПСХ та же, что и при АГ и ПБЦ. Больные АГ и ЯК отвечают на терапию ПР хуже, чем больные с одним АГ (не столь часты ремиссии, чаще и быстрее выявляют прогрессирование к ЦП). Эти пациенты, возможно, должны лечиться УДХК большими дозами (до 15-20 мг/кг/сутки), если признаки холестаза выражены.
При сочетании АГ и ХВГ назначают ПР 20 мг/сутки или 10 мг/сутки ПР и 50 мг/сутки АЗА на три–шесть месяцев, если превалируют признаки АГ. Рекомбинантный ИФН в дозе 3 млн. МЕ/сутки три раза в неделю до 6 месяцев назначают при выявлении признаков ХВГ и маркеров репликации вируса либо неэффективности стероидной терапии [21, 5]. Лечение таких больных представляет собой сложную задачу, так как ПР усиливает вирусную репликацию, а ИФН может усилить иммуноопосредованный печеночно-клеточный некроз, перевести ХВГ в АГ, который до этого мог быть в латентном состоянии, обострить течение болезни с развитием внепеченочных аутоиммунных проявлений, индуцировать выброс антител с неясным клиническим значением. Поэтому лечение состоит в правильном определении преобладания тех или иных клинических синдромов или признаков. В любом случае обострение болезни печени или внезапное появление признаков аутоиммунного заболевания у пациентов с признаками АГ, но с преобладанием вирусного поражения указывает на необходимость прерывания лечения ИФН.
Тактика лечения больных криптогенным ХГ состоит в назначении ПР 10-20 мг/сутки вместе с 50 мг/сутки АЗА до появления ремиссии или максимального эффекта.
Литература
Согласно эпидемиологическим данным Всемирной организации здравоохранения (ВОЗ), на 2017 год в мире насчитывается около 150 миллионов человек, которые были инфицированы вирусом гепатита С. Гепатит С — это опасное вирусное заболевание печени. Этот вирус может приводить к развитию как острой, так и хронической формы гепатита, которая варьируется по тяжести – от легкой болезни, продолжающейся несколько недель, до серьезной пожизненной болезни. При своевременной диагностике и правильно подобранном лечении данная патология полностью излечима, особенно, если не медлить со сдачей анализов на выявления гепатита С в крови. Иначе последствия могут быть трагическими — гепатит С часто приводит к развитию необратимых изменений, потенциально угрожающих для жизни.
Благодаря наличию в своих стенах современного медицинского оборудования, Юсуповская больница занимается диагностикой всех видов вирусных гепатитов. В клиники терапии каждому пациенту окажут не только квалифицированную помощь от специалистов высшей категории с многолетним стажем работы, но и понимание, поддержку в его нелегкой борьбе с ужасным заболеванием.

Гепатит С: что нужно знать пациенту
Гепатит С — один из самых коварных и смертельных вирусов. В 90% случаев протекают бессимптомно. Из-за способности маскировать истинную причину под видом множества других заболеваний, его прозвали «ласковый убийца». Если болезнь все-таки дала о себе знать, активная ее фаза делится на два основных периода: преджелтушный и желтушный. В первом (раннем) периоде отмечаются такие характерные для вирусных инфекций симптомы, как:
- общая слабость;
- кожный зуд;
- расстройства пищеварения: тошнота, рвота, диарея;
- повышение температуры тела до 38°С;
- головные боли, миалгия, артралгия.
На втором (желтушном) периоде, когда поражена печень, в кровь выбрасывает большое количество билирубина — желтого пигмента. Именно благодаря иктеричному окрашиванию кожных покровов пациента становится очевидно, что у него присутствуют явные проблемы с печенью, и назначается комплекс лабораторных исследований крови, мочи и кала.
Однако многие случаи заражения протекают бессимптомно, без характерной клинической картины. После инкубационного периода, который длиться от пары недель до пары месяцев, пациент может и не подозревать, что он является носителем вируса не только в продромальной (преджелтушной) стадии, но и в желтушной — по причине отсутствия ее как таковой. Например, в 2/3 всех случаев гепатит С проходит в атипичной (безжелтушной, или субклинической) форме.
Анализ гепатит С: особенности и подготовка
Всегда кровь на биохимический анализ сдается строго натощак, непосредственно в утренние часы, до полудня. Это обусловлено суточными ритмами в организме человека, которые влияют на содержание гормонов в крови. Количественный анализ на гепатит С (антигены и антитела) сдается в любое время суток, но тоже, обязательно, натощак: важно не принимать пищу в течение 4–6 часов до забора крови. В обоих случаях используется венозная кровь, которая как биоматериал более качественна, чем капиллярная либо артериальная.
Накануне сдачи любых анализов крови рекомендуется избегать чрезмерных физических и психоэмоциональных нагрузок, приема алкоголя и тяжелой пищи. Питьевой режим должен быть обычным.
Количественные и качественные анализы на гепатит С
Этот вид гепатита имеет шесть разновидностей, поэтому анализы необходимо проводить в комплексе. К группе риска относятся люди, принимающие внутривенные наркотики (кокаин, героин), ведущие беспорядочную половую жизнь, медработники (особенно хирурги и анестезиологи), а также пациенты, которым показаны гемодиализ или переливания крови.
В профилактических целях либо при подозрении вирусное поражение печени сдаются следующий качественный и количественный анализ на гепатит С:
- Анализ на anti-HCV-total (антитела к антигенам вируса гепатита C). Качественный анализ. Положительный результат означает инфицирование или период восстановления после перенесенной инфекции. Отрицательный результат - инкубационный период;
- Определение РНК (HCV-RNA) в сыворотке или плазме крови. Может быть качественным и количественным. При качественном анализе результат «обнаружено» подтверждает заражение гепатитом C. Отрицательный результат говорит об отсутствии вируса в крови пациента;
При количественном анализе плазмы крови на гепатит С:
- «не обнаружено»: РНК гепатита С не выявлена;
- 100 000 000 МЕ/мл: результат интерпретируется как: «РНК гепатита С выходит за пределы линейного диапазона, тест ставился в разведении 1: Х».
При количественном анализе сыворотки крови на гепатит С:
- «не обнаружено»: РНК гепатита С не выявлена;
- 2 до 10 8 МЕ/мл: результат положительный;
- 10 8 МЕ/мл: результат положительный, концентрация РНК гепатита С более 108 МЕ/мл.
- Определение антител IgG (recomBlot HCV IgG). Качественный тест. Отрицательный результат говорит об отсутствии инфицирования. Положительный результат - пациент был инфицирован ранее. Сомнительный результат - возможно, была инфекция.
Расшифровка анализа крови на гепатит С
Расшифровка анализа крови на гепатит С и верификация диагноза, с учетом клинической и эпидемиологической картины, под силу только специалисту.
При отрицательном результате в крови, по итогам всех проведенных тестов, не находят никаких маркеров вирусного гепатита. Тогда можно говорить об отсутствии заболевания. Однако в некоторых случаях врачи рекомендуют сделать анализы повторно через две недели.
При положительном результате анализа на гепатит проводится повторный уточняющий анализ через две-три недели. Возможен вариант, когда пациент только что переболел острой формой вирусного гепатита и маркёры в крови пока еще сохраняются.

Анализ на гепатит С: цена, Москва
Стоимость анализа на гепатит С в частных клиниках Москвы варьируется в диапазоне от 500-2000 рублей. Качественный анализ на гепатит С стоит в среднем 700 рублей. Количественный анализ на гепатит С методом ПЦР обойдется жителям Москвы примерно в 2500 рублей.
Юсуповская больница специализируется на диагностике и проведении анализов на выявление всех типов вирусных гепатитов. В стационаре клиники работает собственная современная лаборатория, сотрудники которой постоянно усовершенствуются в своей профессии и проходят специальные курсы по повышению квалификации. В больнице разрабатывают новые медикаментозные методики лечения различных видов вирусных гепатитом, в том числе и гепатит С. Ответственность перед каждым пациентом и высокий уровень профессионализма сотрудников – это визитная карточка Юсуповской больницы. Для записи на прием, звоните по телефону.
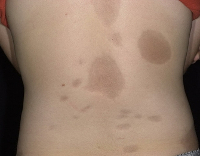
Очаговая склеродермия – это хроническое заболевание соединительной ткани, характеризующееся преимущественным поражением кожных покровов. Клинически проявляется уплотнением (индурацией) различных участков кожи с последующей атрофией и изменением пигментации, образованием контрактур. Диагноз ставится на основании симптоматики, обнаружения в крови антинуклеарного фактора и антицентромерных антител. В сомнительных случаях проводится гистологическое исследование кожи. Лечение заключается в применении глюкокортикостероидов, иммунодепрессантов, антифиброзных средств, блокаторов кальциевых каналов и проведении ПУВА-терапии. В ряде случаев выполняются хирургические операции.
МКБ-10
Общие сведения
Очаговая (локализованная, ограниченная) склеродермия – хроническое аутоиммунное заболевание из группы диффузных болезней соединительной ткани. Патология встречается повсеместно, распространенность составляет от 0,3 до 3 случаев на 100 000 человек. Чаще страдают женщины европеоидной расы. Возраст манифестации очаговой склеродермии зависит от формы. Бляшечная склеродермия чаще встречается у взрослых (30-40 лет), линейная - у детей от 2 до 14 лет, склероатрофический лихен – у женщин старше 50 лет. При локализованной форме, в отличие от системной, поражение внутренних органов в большинстве случаев либо минимально, либо отсутствует. Имеется ассоциация склеродермии с патологиями щитовидной железы (тиреоидитом Хашимото, болезнью де Кервена).
Причины
Точная причина заболевания неизвестна. Предполагается этиологическая роль бактерии Borrelia burgdorferi, вызывающей лайм-боррелиоз, однако убедительных данных за эту теорию на сегодняшний день нет. В развитии склеродермии важную роль играет наследственная предрасположенность. Были выявлены более частые случаи очаговой склеродермии среди близких родственников. При проведении генетических исследований обнаружена взаимосвязь между определенными генами гистосовместимости (HLA – DR1, DR4) и локализованной формой заболевания. Провоцирующими факторами, способствующими возникновению склеродермии, являются переохлаждения, травмы, постоянные вибрационные воздействия на кожу, прием лекарственных препаратов (блеомицина). Триггерными эффектами также обладают различные химические соединения (хлорвинил, кремний, нефтепродукты, сицилий, эпоксидная смола, пестициды, органические растворители).
Патогенез
Выделяют три основных патогенетических механизма склеродермии – фиброз (разрастание соединительной ткани), аутоиммунное повреждение и сосудистые нарушения. Иммунная аутоагрессия заключается в выработке лимфоцитами антител к соединительной ткани и ее компонентам. Также лимфоциты синтезируют интерлейкины, которые стимулируют пролиферацию фибробластов, гладкомышечных клеток и образование коллагена. Разрастающаяся при этом соединительная ткань замещает нормально функционирующую ткань. В результате повреждения эндотелия сосудов антителами и пролиферирующими гладкомышечными клетками снижается уровень простациклина (вещества, обладающего антиагрегантными и вазодилатирующими свойствами). Это приводит к спазму микрососудов, повышению адгезии и агрегации форменных элементов крови, внутрисосудистой коагуляции и микротромбозу.
Классификация
Очаговая склеродермия подразделяется на множество форм. Наиболее распространенными являются бляшечная и линейная. У ряда пациентов могут наблюдаться одновременно несколько вариантов заболевания. Существует целый ряд классификаций, но наиболее оптимальной и широко используемой считается классификация клиники Мэйо, включающей следующие разновидности очаговой склеродермии:
- Бляшечная. Данная форма в свою очередь подразделяется на поверхностную (морфеа) и узловатую (келоидоподобную). Характерны типичные участки уплотнения кожи с атрофией и нарушением пигментации.
- Линейная. К ней относятся полосовидная, саблевидная формы, а также прогрессирующая гемиатрофия лица Парри-Ромберга. Очаги располагаются в виде линий по ходу сосудисто-нервного пучка.
- Генерализованная (многоочаговая). Проявляется сочетанием бляшечного и линейного вариантов. Очаги распространены по всему телу.
- Буллезная. При данной разновидности на коже возникают пузыри с жидкостным содержимым, оставляющие после себя эрозии.
- Пансклеротическая инвалидизирующая. Наиболее неблагоприятная форма очаговой склеродермии. Характеризуется тяжелым, прогрессирующим течением, плохо поддается лечению. Поражаются все слои кожи и ткани, лежащие под ней. Развиваются грубые контрактуры суставов и длительно незаживающие язвы на коже.
- Склероатрофический лихен Цумбуша (болезнь белых пятен). Характерно образование пятен белого цвета, сопровождающихся нестерпимым зудом. Преимущественная локализация пятен – половые органы.
Симптомы
Для клинической картины типично образование на коже очагов, которые проходят три последовательных стадий развития – отек, индурацию (уплотнение) и атрофию. В начале заболевания на коже конечностей, шеи или туловища появляются пятна сиреневого или лилового цвета, имеющие нечеткие края. Размер пятен может сильно варьировать – от просяного зерна до размеров ладони и больше. На этом этапе пациент не испытывает каких-либо неприятных ощущений или боли. Затем пятна начинают отекать, кожа в центре очага уплотняется, становится блестящей, приобретает цвет слоновой кости. Пациент начинает ощущать зуд, покалывания, стянутость кожи, болезненность. Далее наступает стадия атрофии. Кожа в очагах истончается, прекращается рост волос, нарушается потоотделение, возникает стойкая дисхромия (гипер- или депигментация) и телеангиэктазии. Иногда развивается атрофодермия (участки западения кожи).
При линейной склеродермии очаги расположены по ходу нервов и сосудов. В случае локализации на коже лица очаги по внешнему виду напоминают рубец от удара саблей (саблевидная форма). Прогрессирующая гемиатрофия представляет собой глубокий процесс с поражением всех тканей половины лица - кожи, подкожной клетчатки, мышц и костей лицевого скелета, что приводит к выраженной деформации лица, обезображивающей внешний вид пациента. Также происходит атрофия половины языка и снижение вкусовой чувствительности.
Из внекожных признаков очаговой склеродермии стоит отметить офтальмологические и неврологические проявления при гемиатрофии Парри-Ромберга. Они включают выпадение ресниц и бровей на стороне поражения, западение глазного яблока из-за атрофии глазных мышц и орбитальной клетчатки, нейропаралитический кератит, головокружения, когнитивные нарушения, мигренозные головные боли, эпилептические припадки. Также возможно развитие феномена Рейно. Симптомы синдрома Рейно следующие – стадийное изменение окраски кожи пальцев рук вследствие вазоспазма и последующей гиперемии (бледность, цианоз, покраснение), сопровождающееся онемением, болью и покалыванием в пальцах рук. Остальные экстрадермальные проявления, характерные для системной склеродермии, встречаются крайне редко.
Осложнения
Наиболее распространенная проблема рассматриваемого заболевания – косметические дефекты. Серьезные осложнения, представляющие угрозу для жизни больного, возникают редко. К ним относятся нарушение мозгового кровообращения при гемиатрофии лица, ишемия и гангрена пальцев рук при феномене Рейно, выраженные контрактуры суставов, инвалидизирующие пациента. Через несколько лет после дебюта болезни могут развиться тяжелые поражения внутренних органов – фиброз легких, легочная гипертензия, фиброз миокарда, перикардит, стриктуры пищевода, острая нефропатия, почечная недостаточность.
Диагностика
Пациентов с очаговой склеродермией курируют врачи ревматологи и дерматологи. При постановке диагноза учитывается клиническая картина, семейный анамнез. Все методы диагностики направлены в первую очередь на определение степени вовлечения внутренних органов и исключение системной склеродермии. С этой целью применяются следующие исследования:
- Лабораторные. В анализах крови выявляются эозинофилия, повышение уровня ревматоидного фактора, гаммаглобулинов, высокие титры антицентромерных антител и антинуклеарного фактора (АНФ). Наличие антител к топоизомеразе (анти-Scl 70) свидетельствует в пользу системного процесса. При развитии «склеродермической почки» в моче появляются белок и эритроциты.
- Инструментальные. При капилляроскопии наблюдается дилатация капилляров без участков некроза. По данным ФЭГДС могут встречаться признаки эзофагита, стриктуры пищевода. При фиброзе миокарда на ЭКГ иногда обнаруживаются нарушения ритма сердца, на ЭхоКГ – зоны гипокинеза, выпот в перикардиальную полость. На рентгенографии или компьютерной томографии легких отмечаются интерстициальные изменения.
- Гистологическое исследование биоптата кожи. Заключительный этап, позволяющий достоверно поставить диагноз. Проводится при сомнительных результатах предыдущих исследований. Характерны следующие признаки - инфильтрация лимфоцитами, плазмоцитами и эозинофилами в ретикулярном слое дермы, утолщенные коллагеновые пучки, набухание и склероз сосудистой стенки, атрофия эпидермиса, сальных и потовых желез.
Очаговую склеродермию дифференцируют с другими формами склеродермии (системной, склеродермой Бушке), дерматологическим заболеваниями (саркоидозом кожи, липонекробиозом, склеродермоподобной формы поздней кожной порфирии, базально-клеточным раком), поражением мягких тканей (панникулитом, липодерматосклерозом, эозинофильным фасциитом). В дифференциальной диагностике принимают участие онкологи, гематологи.
Лечение
Этиотропной терапии не существует. Метод лечения и вид лекарственного средства необходимо подбирать с учетом формы заболевания, тяжести течения и локализации очагов. При линейной и бляшечной формах используются топические глюкокортикостероиды высокой и сверхвысокой активности (бетаметазон, триамцинолон), синтетические аналоги витамина Д. При выраженной индурации кожи эффективны аппликации с диметилсульфоксидом. В случае поражений внутренних органов с целью уменьшения фиброзообразования назначаются пеницилламин и инъекции гиалуронидазы.
При неглубоких процессах хорошим терапевтическим действием обладает ПУВА-терапия, которая включает облучение кожи ультрафиолетовыми волнами длинного спектра с одновременным пероральным или наружным применением фотосенсибилизаторов. Тяжелое поражение кожи служит показанием к применению иммунодепрессантов (метотрексата, такролимуса, микофенолата), синдром Рейно - блокаторов кальциевых каналов (нифедипина) и препаратов, улучшающих микроциркуляцию (пентоксифиллина, ксантинола никотината). При склероатрофическом лихене проводится низкоинтенсивная лазеротерапия. В случае развития контрактур суставов, значительно затрудняющих движения, или грубых деформаций скелета и косметических дефектов лица требуется хирургическая операция.
Профилактика и прогноз
В подавляющем большинстве случаев очаговая склеродермия имеет доброкачественное течение. Правильно подобранная терапия позволяет добиться регресса симптомов. Иногда наступают спонтанные ремиссии заболевания. Неблагоприятные исходы возникают при тяжелых формах (прогрессирующей гемиатрофии лица, пансклеротической инвалидизирующей склеродермии), а также поражении внутренних органов. Эффективных методов профилактики не разработано. Рекомендуется избегать или максимально ограничить контакт кожи с химическими соединениями (кремнием, сицилием, хлорвинилом, нефтепродуктами, органическими растворителями, пестицидами, эпоксидной смолой).
2. Ревматические заболевания/ Под ред. Дж.Х. Клиппела, Дж.Х. Стоуна, Л.Дж. Кроффорд, П.Х. Уайт – 2012.
3. Диффузные болезни соединительной ткани: руководство для врачей/ под ред. проф. Мазурова В.И. –2009.
Читайте также: