
Амилоидоз кожи что это такое
Обновлено: 25.04.2024
Амилоидоз – это системное заболевание, характеризующееся отложением в тканях амилоида (сложного белково-полисахаридного комплекса).
Амилоидоз приводит к атрофии, склерозу и недостаточности различных органов.
Частота встречаемости – не менее 1:50 000 (в основном возникает после 60 лет).
1. Первичный амилоидоз обусловлен изменениями клеток при миеломной болезни, моноклональной гипергаммаглобулинемии, макроглобулинемии Вальденстрема.
Амилоид состоит из легких цепей иммуноглобулинов, их синтез резко увеличен при указанных заболеваниях.
2. Вторичный амилоидоз возникает вследствие хронических воспалительных заболеваний (например, при ревматоидном артрите, остеомиелите, бронхоэктатической болезни, малярии, туберкулезе, лепре).
Амилоид состоит из фибриллярного белка амилоида и продуктов его распада – сывороточного амилоидного белка.
3. Семейный (идиопатический) амилоидоз. Обычно – врожденный дефект ферментов. Имеется несколько форм врожденного амилоидоза, например при средиземноморской лихорадке (семейный пароксизмальный полисерозит – заболевание неясной причины, проявляющееся болями в животе, приступами лихорадки, плевритом, артритом и высыпаниями на коже).
4. Старческий амилоидоз.
5. Диализный амилоидоз развивается при проведении гемодиализа.
Причины преимущественного поражения тех или иных органов (почек, кишечника, кожи) неизвестны.
Признаки и течение заболевания разнообразны и зависят от локализации амилоидных отложений, степени их распространенности в органах, длительности заболевания, наличия осложнений.
Чаще наблюдают комплекс симптомов, связанных с поражением нескольких органов.
*желудочно-кишечного тракта
- Увеличение размеров языка
- Нарушение глотания или запоры
- Опухолевидные отложения амилоида в желудке или кишечнике (не часто)
Амилоидоз пищевода обычно сопровождается поражением других отделов пищеварительной системы. Характерны затруднение глотания при проглатывании плотной и сухой пищи, отрыжка.
Амилоидоз желудка обычно сочетается с амилоидозом кишечника и других органов. Проявления: ощущение тяжести в подложечной области после еды, изжога, отрыжка, тошнота.
Амилоидоз кишечника возникает часто, проявляется ощущением дискомфорта, тяжести, реже умеренными болями в животе, нарушениями стула (запорами или упорной диареей). Изолированный опухолевидный амилоидоз кишечника протекает под маской опухоли (боль, непроходимость кишечника), и обычно его выявляют во время операции.
Амилоидоз печени наблюдают сравнительно часто, характерны увеличение и уплотнение печени, нередко появляются синдром портальной гипертензии, реже возникают боли в правом подреберье, тошнота, отрыжка, желтуха, кровоизлияния.
Амилоидоз поджелудочной железы обычно протекает под маской хронического панкреатита; характерны тупая боль в левом подреберье, отрыжка, тошнота, рвота.
- Устойчивая к лечению застойная сердечная недостаточность
- Нарушения ритма и проводимости
- Очаговые поражения миокарда (псевдоинфаркт).
*нервной системы
- Периферическая полиневропатия (ощущение жжения, покалывания, «мурашек» в конечностях, нарушение чувствительности)
- Нарушения вегетативной нервной системы (головные боли, голокружение, потливость)
- Ортостатическая гипотензия
- Импотенция
- Сфинктерные расстройства (недержание мочи, кала).
*сухожилий, хрящей
- Синдром запястного канала (онемение, покалывание и пульсирующая боль в пальцах)
- Симметричный полиартрит, плечелопаточный периартрит, плотный отек околосуставных.
*органов дыхания
- Охриплость голоса
- Бронхит
- Опухолевидный легочный амилоидоз.
Поражения кожи – папулы, бляшки, узлы, кровоизлияния вокруг глаз («симптом очков»).
Наследственный амилоидоз – группа заболеваний, которые характеризуются накоплением нерастворимых белковых комплексов (амилоида) в различных тканях – главным образом, в нервной и пищеварительной системах, почках и миокарде. Симптомами являются неврологические нарушения, патологии сердца (кардиомиопатия), нефропатия и диспепсические расстройства. При различных формах наследственного амилоидоза превалирует поражение определенной системы. Диагностика производится на основании клинической картины и гистологического изучения тканей, для некоторых форм разработаны методы молекулярно-генетического определения. Лечение включает использование иммуносупрессивных препаратов, поддерживающую и симптоматическую терапию.
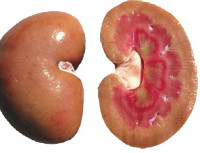
Общие сведения
Наследственный или семейный амилоидоз – генетически обусловленное нарушение обмена протеинов, при котором происходит образование аномального белка с его последующим осаждением в составе иммунных комплексов в различных тканях. Как нозологическая единица амилоидоз известен давно, но генетическая природа некоторых форм этого состояния была обнаружена только в последние десятилетия. Дифференцировка наследственных и ненаследственных форм значительно осложняется тем, что ряд генетически обусловленных патологий (например, периодическая болезнь) также характеризуются отложением амилоида, но сейчас принято считать, что белковые нарушения при таких заболеваниях имеют вторичный характер.
В настоящий момент к первичному наследственному амилоидозу достоверно относят только типы патологии, обусловленные мутацией гена TTR. Дискуссионным вопросом является классификация так называемого амилоидоза финского типа – установлено, что это заболевание обусловлено дефектом гена GSN, однако до сих пор неясно, являются ли отложения амилоида при этом первичным нарушением или же следствием каких-либо иных процессов. Некоторые врачи-генетики ассоциируют этот тип заболевания с наследственным амилоидозом 4-го типа, но имеющим другую генетическую природу. Встречаемость наследственного амилоидоза не определена по причине редкости этого состояния.
Причины наследственного амилоидоза
Патогенез наследственного амилоидоза связан с нарушением стабильности молекул транстиретина – по причине мутации гена TTR изменяется структура этой белковой молекулы и она становится нестабильной. В ряде случаев нестабильность незначительно влияет на ее функции, но иногда конформация молекулы изменяется настолько, что она приобретает иммуногенные свойства. Аномальный протеин вызывает реакцию, сходную с аллергической (иммунокомплексный тип), в результате чего возникают нерастворимые белковые комплексы, склонные к отложению в экстрацеллюлярном пространстве тканей. Клинические проявления наследственного амилоидоза зависят от того, где преимущественно происходит отложение протеиновых комплексов, которые вызывают нарушения функциональности органов.
Классификация и симптомы наследственного амилоидоза
Наследственный амилоидоз, обусловленный мутациями гена TTR, способен проявляться нарушениями со стороны разных органов и тканей – нервов, желудка и 12-типерстной кишки, почек, сердца, глаз. При этом клиническая картина заболевания зависит от характера мутации, различные формы отличаются между собой превалирующим поражением каких-либо органов, выраженностью нарушений и длительностью течения. На сегодняшний день выделяют 6 основных фенотипических форм наследственного амилоидоза.
Тип 1 (синдром Андраде) – является наиболее распространенным вариантом наследственного амилоидоза, впервые был описан в 1952-м году. Наиболее распространен в Португалии, а также в бывших португальских колониях (Латинской Америке). Начало этой формы наследственного амилоидоза чаще всего регистрируется в возрасте 30-40 лет, первым симптомом является нарушение чувствительности (болевой, температурной, тактильной) на коже нижних конечностей. Полиневропатия постепенно становится все более выраженной, помимо поражения чувствительных нервов со временем присоединяются двигательные расстройства мышц дистальных отделов ног. Из-за нарушения иннервации наследственный амилоидоз часто приводит к трофическим язвам нижних конечностей. Патология затрагивает и вегетативную нервную систему – у больных наблюдаются нарушения потенции и ортостатическая гипотония. На конечных этапах развития наследственного амилоидоза 1-го типа отложения белка возникают в почках, сердце и роговице, вызывая нарушения в этих органах и приводя к летальному исходу – чаще всего, через 10-13 лет после появления первых симптомов.
Тип 2 (синдром Рукавина) – очень редкий тип наследственного амилоидоза, который достоверно был описан лишь у представителей двух семей в США в 1956-м году. Он проявляется в 35-45 лет и начинается с выраженного туннельного синдрома запястного канала, к которому через несколько лет присоединяются поражение чувствительных и вегетативных нервов. Этот тип наследственного амилоидоза прогрессирует очень медленно, через 15-20 лет после начала заболевания могут отмечаться отложения амилоида в кишечнике, сердце, почках.
Тип 3 (синдром Ван Аллена) – разновидность наследственного амилоидоза (наследственной амилоидной невропатии), характеризующаяся превалирующим поражением нервов и кишечника. Клинические проявления этого заболевания очень схожи с 1-м типом, основное отличие заключается в отсутствии значительного поражения вегетативной нервной системы. Также у больных этой формой наследственного амилоидоза часто отмечаются гастриты и язвы 12-типерстной кишки.
Тип 4 или наследственный амилоидоз финского типа был впервые описан в 1971-м году, по поводу его генетической природы, как уже было сказано выше, в настоящий момент ведется активная дискуссия. В основном при этой форме заболевания поражаются черепно-мозговые нервы, нарушается чувствительность верхних конечностей. Со временем возникает решетчатая атрофия роговицы и снижается тургор кожи, на заключительных этапах развития патологии возникают выраженная полинейропатия и отложения амилоида в сердце. В отличие от других форм наследственного амилоидоза, этот тип может наследоваться по аутосомно-рецессивному механизму (в том случае, если заболевание обусловлено мутациями гена GSN).
Наследственный кардиомиопатический амилоидоз или наследственный амилоидоз датского типа проявляется слабо выраженными неврологическими нарушениями и значительным поражением миокарда (амилоидоз сердца). Отложения амилоида приводят к быстрому развитию кардиомиопатии и сердечной недостаточности, что становится причиной летального исхода.
Нефропатический амилоидоз с глухотой, крапивницей и лихорадкой – редкая, но достаточно тяжелая форма наследственного амилоидоза, при котором у больных в возрасте 20-30 лет возникают лихорадка неустановленного генеза и частые приступы крапивницы. Затем к этим проявлениям присоединяется нейросенсорная тугоухость, которая в последующем осложняется полной глухотой. Поражение почек (амилоидоз почек) нередко приводит к хронической почечной недостаточности, что и становится причиной смерти от уремии большинства больных.
Диагностика и лечение наследственного амилоидоза
Диагностика наследственного амилоидоза производится на основании данных осмотра пациента, общеклинических анализов, оценки состояния ключевых органов (нервной системы, сердца, почек), гистологического изучения тканей, молекулярно-генетических исследований. При осмотре могут выявляться признаки неврологических поражений (нарушения чувствительности и двигательных функций, гипо- или арефлексия), в запущенных случаях отмечается атрофия мышц, обусловленная отсутствием иннервации. Офтальмологическое обследование при наследственном амилоидозе нередко обнаруживает признаки отложения амилоида в стекловидном теле, возможна атрофия или изъязвление роговицы. В ряде случаев могут отмечаться нарушения со стороны кожных покровов – снижение тургора, крапивница, трофические язвы.
Ультразвуковое исследование сердца и почек при наследственном амилоидозе также нередко дает немало информации об этом заболевании. В случае отложения амилоида в почках наблюдается их увеличение и уплотнение структуры, иногда определяется наличие кист. На конечных этапах развития амилоидоза может возникать уменьшение почек по причине их атрофии. На ЭКГ уменьшается амплитуда и вольтаж всех зубцов, ЭхоКГ обнаруживает значительное увеличение толщины миокарда и размеров левого желудочка. В ряде случаев методом УЗИ при наследственном амилоидозе можно выявить отложения амилоида и в других органах – печени, селезенке, крупных сосудах. При биопсии пораженных органов больных, страдающих наследственным амилоидозом, отмечается типичная для этого заболевания гистологическая картина – отложение эозинофильных масс с концентрацией вокруг кровеносных сосудов. Современная генетика способна выявлять мутации в гене TTR методом прямого секвенирования.
Специфического лечения наследственного амилоидоза не существует, в основном применяется симптоматическая и поддерживающая терапия. Для замедления течения заболевания используют иммуносупрессивные средства (в частности – кортикостероиды) изредка назначают цитотоксические препараты. В случае значительного поражения почек по причине наследственного амилоидоза показан гемодиализ. Симптоматическое лечение также необходимо при кардиомиопатии и сердечной недостаточности. Рекомендуется принимать повышенные дозировки витаминов группы В, так как они замедляют развитие нейропатии и способствуют улучшению общего состояния больного.
Прогноз наследственного амилоидоза
В большинстве случаев прогноз наследственного амилоидоза неблагоприятный, так как многие формы этого заболевания приводят к летальному исходу через 10-25 лет после его начала. Симптоматическая и поддерживающая терапия, гемодиализ, применение иммуносупрессоров способны замедлить развитие этой патологии. С учетом того, что чаще всего симптомы наследственного амилоидоза проявляются в 30-40 лет, своевременная диагностика заболевания и разумная поддерживающая терапия способны обеспечить выживаемость больных до старости без значительного ухудшения качества жизни.
Амилоидоз (амилоидная дистрофия) - нарушение белков о го обмена, сопровождающееся образованием и отложением в тканях специфического белково-полисахаридного комплекса - амилоида. Амилоид долгое время персистирует в организме и даже после смерти в течение долгого времени не подвергается гниению (И. В. Давыдовский, 1967).
Что провоцирует / Причины Амилоидоза (амилоидной дистрофии):
Развитие амилоидоза связано с нарушением белково-синтетической функции ретикуло-эндотелиальной системы, накоплением в плазме крови аномальных белков, служащих аутоантигенами и вызывающих образование аутоантител. В результате взаимодействия антигена с антителом происходит осаждение грубодисперсных белков, участвующих в образовании амилоида. Откладываясь в тканях (например, в стенках сосудов, железистых и т. п.), амилоид вытесняет функционально специализированные элементы органа, что ведёт к гибели этого органа.
Патогенез (что происходит?) во время Амилоидоза (амилоидной дистрофии):
AL-амилоидоз (immunoglobulin light chains derived) - первичный амилоидоз, вызванный накоплением в плазме крови аномальных легких цепей иммуноглобулинов, синтезируемых малигнизированными В-лимфоцитами при миеломной болезни (болезнь Рустицкого-Калера)
AA-амилоидоз (acquired) - вторичный амилоидоз, вызванный гиперсекрецией печенью белка острой фазы альфа-глобулина в ответ на хроническое (чаще гнойное) воспаление. Примером таких воспалений служит лепра, бронхоэктатическая болезнь, туберкулез и т. д. Подвидом данного амилоидоза является ASC-амилоидоз (systemic cardiovascular), развивающийся у лиц свыше 90 лет. Причины патогенеза неизвестны.
AF-амилоидоз (средиземноморская перемежающая лихорадка) - наследственная форма амилоидоза, с аутосомно-рецессивным механизмом передачи. Данным видом амилоидоза страдают люди, принадлежащие к определенным этническим группам, живущим по побережью Средиземного моря (евреи-сефарды, греки, арабы, армяне). Существуют разновидности амилоидоза, характерные для определенной географической местности: «португальский амилоидоз» (с преимущественным поражением нервов нижних конечностей), «американский амилоидоз» (с преимущественным поражением нервов верхних конечностей), семейный нефропатический амилоидоз, или «английский амилоидоз», протекающий с симптомами крапивницы, глухоты и лихорадки.
AH-амилоидоз (hemodialisis-related) - наблюдается исключительно у больных, находящихся на гемодиализном лечении. Патогенез связан с тем, что, в норме утилизирующийся почками бета-2-микроглобулин MHC I класса, не фильтруется в гемодиализаторе и накапливается в организме.
AE-амилоидоз - форма местного амилоидоза, развивающаяся в некоторых опухолях, например, в медуллярном раке C-клеток щитовидной железы. В этом случае предшественником амилоида являются патологические фрагменты кальцитонина.
Амилоидоз финского типа - редкий тип амилоидоза, вызываемый мутацией гена GSN , кодирующего белок джелсолин .
Симптомы Амилоидоза (амилоидной дистрофии):
Интенсивность клинических проявлений зависит от локализации и распространенности амилоидных отложений. Этим обуславливаются длительность заболевания и наличие осложнений. Наиболее типично поражение почек, иногда бывают поражения пищевода, селезенки, кишечника, а также желудка. При амилоидозе почек наблюдается длительный скрытый период болезни, при котором отсутствуют значительные симптомы, - наблюдаются лишь незначительная слабость и снижение активности. После двух недель течения скрытого периода появляются отеки почек, их дисфункция (появление в моче белков - развитие протеинурии, а также форменных элементов крови), сердечная недостаточность. При амилоидозе желудка наблюдаются тяжесть в эпигастральной области, ослабление перистальтики желудка после употребления пищи. При амилоидозе кишечника наблюдаются тяжесть и тупые боли в области живота, диарея.
Диагностика Амилоидоза (амилоидной дистрофии):
- В крови анемия, лейкоцитоз, повышение СОЭ, почти в 80% случаев в начале заболевания возникают снижение количества белков, гиперглобулинемия, снижения натрия, тромбоцитов, кальция. При поражении печени - повышение холестерина, в ряде случаев - повышение билирубина, повышение активности щелочной фосфатазы
- Оценка функций щитовидной железы - возможено снижение функций щитовидной железы
- Оценка функций почек - почти в 50% случаев амилоидоз начинается с почечной недостаточности. При исследовании мочи, помимо белка, в осадке обнаруживают цилиндры, эритроциты, лейкоциты
- При первичном амилоидозе в плазме крови и/или моче обнаруживают увеличение содержания амилоида
- При вторичном амилоидозе следует обратить внимание на лабораторные признаки хронических воспалительных заболеваний
- В кале - большое количество жира, крахмала, мышечных волокон
- Эхокардиография (при подозрении на поражение сердца)
- Рентгенологическое исследование
- Функциональные клинические пробы с конго красным и метиленовым синим (быстрое исчезновение красителей при внутривенном введении из сыворотки крови вследствие их фиксации амилоидом и значительное снижение выделения их почками). При первичном амилоидозе эти пробы не всегда информативны
- Биопсия органов - наиболее информативный метод.
Лечение Амилоидоза (амилоидной дистрофии):
Общие принципы лечения амилоидоза
- Режим домашний, за исключением тяжёлых состояний (выраженная сердечная недостаточность, хроническая почечная недостаточность)
- Больным амилоидозом показан длительный (1,5-2 года) приём сырой печени (по 100-120 г/сут)
- Ограничение потребления белка, соли пациентам с хронической почечной недостаточностью
- Ограничение соли пациентам с сердечной недостаточностью
- При вторичном амилоидозе - лечение основного заболевания (туберкулёз, остеомиелит, эмпиема плевры и др.), после излечения которого нередко исчезают и проявления амилоидоза
- Перевод пациента с диализным амилоидозом на перитонеальный диализ
- При амилоидозе кишечника, протекающем с упорной диареей, - вяжущие средства (висмута субнитрат, адсорбенты)
- При первичном амилоидозе в начальных стадиях процесса - хлорохин 0,25 г 1 р/сут длительно, сочетание мелфалана и преднизолона, мелфалана, преднизолона и колхицина или только колхицин
- При вторичном амилоидозе - специфическое лечение основного заболевания
- При семейном амилоидозе - колхицин (по 0,6 мг 2-3 р/сут)
- Симптоматическая терапия: витамины, диуретические средства, препараты, снижающие давление, переливание плазмы и т.д.
Хирургическое лечение амилоидоза
- Удаление селезенки может улучшить состояние вследствие уменьшения количества амилоида, образующегося в организме
К каким докторам следует обращаться если у Вас Амилоидоз (амилоидная дистрофия):
Вас что-то беспокоит? Вы хотите узнать более детальную информацию о Амилоидоза (амилоидной дистрофии), ее причинах, симптомах, методах лечения и профилактики, ходе течения болезни и соблюдении диеты после нее? Или же Вам необходим осмотр? Вы можете записаться на прием к доктору .
Заболевание чаще встречается у женщин, чем у мужчин.Возраст заболевших вариабелен от 16 до 59 лет (в среднем 34 года).Заболевания преимущественно наблюдается у лиц с высоким фототипом кожи (IV и V), реже III, и жителей Азии, Ближнего Востока и Южной Америки (редко в странах Европы и Северной Америки).В большинство случаев заболевание носит спорадический характер, но в 10% случаев сообщалось о семейной истории.
Этиология и патогенез не изучены.Предполагается роль хронического раздражения участков кожи (трение, ношение тесной одежды - из за чего заболевание именуют меланозом от полотенец, фрикционным дерматитом от одежды с нейлоном) , хронического ультрафиолетового облучения, женских половых гормонов и атопии.В последнее время имеются исследования, показывающие возможную ассоциацию заболевания с вирусом Эпштейна-Барра и парестетической ноталгией.
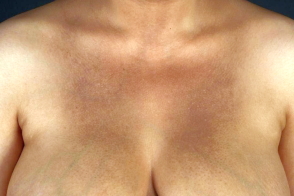
Заболевание характеризуется появлением пятен 2-3 мм в диаметре, от коричневого до красно-коричневого цвета, склонных к слиянию в крупные очаги сетчатой или диффузной гиперпигментации с плохо очерченной границей.В некоторых случаях одновременно с пигментными пятнами наблюдаются мелкие папулы телесного цвета (амилоидный лихен) и гипопигментные участки (пойкилодермический амилоидоз).
Наиболее частая локализация- верхняя часть спины, межлопаточная область, голени и предплечья, реже поражаются ключицы, грудь, лицо, шея и подмышечная впадина.Описан случай появления высыпаний в периорбитальной области.
Субъективно отмечается зуд различной интенсивности, но 20% случаев он отсутствует.Течение заболевания персистирующее, высыпания могут сохранятся неопределенное время.
Диагноз ставиться на основании анамнеза, клинической картины и гистологического исследования, при котором амилоид обнаруживается в сосочковом слое дермы.
- Поствоспалительная гиперпигментация
- Невус Ито
- Пепельный дерматоз
- Черный акантоз
- Парестетическая ноталгия
Эффективного лечения нет, в большинстве случаев заболевание рецидивирует после курса терапии.Предлагаются следующие методы:

Весь контент iLive проверяется медицинскими экспертами, чтобы обеспечить максимально возможную точность и соответствие фактам.
У нас есть строгие правила по выбору источников информации и мы ссылаемся только на авторитетные сайты, академические исследовательские институты и, по возможности, доказанные медицинские исследования. Обратите внимание, что цифры в скобках ([1], [2] и т. д.) являются интерактивными ссылками на такие исследования.
Если вы считаете, что какой-либо из наших материалов является неточным, устаревшим или иным образом сомнительным, выберите его и нажмите Ctrl + Enter.
Амилоидоз - нарушение белкового обмена, сопровождающееся образованием в тканях специфического белково-полисахаридного комплекса (амилоида) и поражением многих органов и систем.
Код по МКБ-10
- Е85 Амилоидоз.
- Е85.0 Наследственный семейный амилоидоз без невропатии.
- Е85.1 Невротический наследственный семейный амилоидоз.
- Е85.2 Наследственный семейный амилоидоз неуточнённый.
- Е85.3 Вторичный системный амилоидоз.
- Е85.4 Ограниченный амилоидоз.
- Е85.8 Другие формы амилоидоза.
- Е85.9 Амилоидоз неуточнённый.
Код по МКБ-10
Эпидемиология амилоидоза
Частота заболевания среди мужчин и женщин при первичном амилоидозе одинакова. Возраст начала болезни колеблется от 17 до 60 лет, а длительность болезни - от нескольких месяцев до 23 лет. Сроки начала заболевания трудно установить, так как первые клинические проявления не соответствуют началу отложения амилоида.

[1], [2], [3], [4], [5], [6], [7], [8], [9], [10], [11], [12], [13], [14]
Причины и патогенез амилоидоза
В зависимости от этиологии и особенностей патогенеза выделяют идиопатический (первичный), приобретённый (вторичный), наследственный (генетический), локальный амилоидоз, амилоидоз при миеломной болезни и APUD-амилоидоз. Наиболее часто встречается вторичный амилоидоз, который по происхождению приближается к неспецифическим (в частности иммунным) реакциям. Он развивается при ревматоидном артрите, анкилозирующем спондилите, туберкулёзе, хроническом остеомиелите, бронхоэктатической болезни, реже при лимфогранулематозе, опухолях почки, лёгкого и других органов, сифилисе, неспецифическом язвенном колите, болезнях Крона и Уиппла, подостром инфекционном эндокардите, псориазе и др.
[15], [16], [17], [18], [19], [20], [21], [22], [23], [24], [25]
Симптомы амилоидоза
Клинические проявления амилоидоза разнообразны и зависят от локализации амилоидных отложений, их распространённости. Локализованные формы амилоидоза, например амилоидоз кожи, долго протекают бессимптомно, как и старческий амилоидоз, при котором отложения амилоида в мозге, поджелудочной железе, сердце нередко обнаруживают только на вскрытии.
Что беспокоит?
Классификация амилоидоза
Согласно классификации Номенклатурного комитета Международного союза иммунологических обществ (Бюллетень ВОЗ, 1993) выделяют пять форм амилоидоза.
- AL-амилоидоз (А - amyloidosis, амилоидоз, L - light chains, лёгкие цепи) - первичный, связанный с миеломной болезнью (амилоидоз регистрируют в 10-20% случаев миеломной болезни).
- АА-амилоидоз (acquired amyloidosis, приобретённый амилоидоз) - вторичный амилоидоз на фоне хронических воспалительных, ревматических, заболеваниях, а также при средиземноморской семейной лихорадке (периодической болезни).
- ATTR-амилоидоз (А - amyloidosis, амилоидоз, TTR - transthyretin, транстиретин) - наследственно-семейный амилоидоз (семейная амилоидная полинейропатия) и старческий системный амилоидоз.
- Аβ2М-амилоидоз (А - amyloidosis, амилоидоз, β2М - β2-микроглобулин) - амилоидоз у больных, находящихся на плановом гемодиализе.
- Локализованный амилоидоз чаще развивается у людей старческого возраста (AIAPP-амилоидоз - при инсулиннезависимом сахарном диабете, АВ-амилоидоз - при болезни Альцгеймера, AANF-амилоидоз - старческий амилоидоз предсердий).
[26], [27], [28], [29], [30], [31], [32]
Диагностика амилоидоза
Амилоидоз следует подозревать при нефропатии, стойкой тяжёлой сердечной недостаточности, синдроме мальабсорбции или полиневропатии неясной этиологии. При нефротическом синдроме или хронической почечной недостаточности необходимо, помимо гломеруло-нефрита, исключить и амилоидоз. Вероятность амилоидоза увеличивается при гепато- и спленомегалии.
[33], [34], [35], [36], [37], [38], [39]
Читайте также: