
Что такое мозоль головного мозга
Обновлено: 17.05.2024
Агенезия мозолистого тела у плода. Диагностика и прогноз при агенезии мозолистого тела плода
Агенезия мозолистого тела является пороком, частота встречаемости которого и клиническая значимость неизвестны. По данным различных исследований частота его выявления варьирует и зависит от особенностей исследуемой популяции и методов диагностики.
Агенезия мозолистого тела также является частью многих менделирующих синдромов. Высокая частота выявления сочетанных пороков при этом состоянии свидетельствует о том, что агенезия мозолистого тела является частью многих широко распространенных нарушений развития.
По данным одного из антенатальных исследований сопутствующие анатомические дефекты обнаруживались в 50% случаев и были, в основном, представлены синдромом Денди-Уокера (Dendy-Walker) и пороками сердца. Аномалии кариотипа (трисомии 18 и 8) были выявлены в 20% наблюдений.
Агенезия мозолистого тела является пороком который сопровождается минимальными анатомическими изменениями, в связи с чем его диагностика особенно до 20 нед беременности, бывает трудна даже для опытных специалистов. Развитие мозолистого тела происходит на поздних этапах церебрального онтогенеза плода, а именно: между 12 и 18 нед гестации, поэтому, вероятно, до 18 нед в большинстве случаев диагноз установить бывает невозможно.
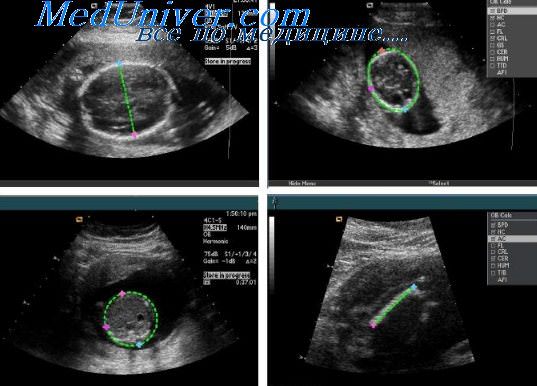
При плановых обследованиях, выполняемых позже этого срока, отсутствие возможности визуализировать полость прозрачной перегородки или расширение атриума боковых желудочков должно наводить на мысль о возможном наличии агене-зии мозолистого тела.
При подозрении на наличие этой аномалии необходимо провести поиск более специфичных признаков. Непосредственная визуализация отсутствия мозолистого тела возможна при использовании срединных фронтальной (коронарной) и продольной (сагиттальной) плоскостей сканирования. Изображения в этих плоскостях не всегда бывает легко получить, особенно при теменном предлежании плода. В таких случаях большие преимущества имеет трансвагинальная эхография.
Агенезия мозолистого тела может быть полной или частичной. В последнем случае, который называется дисгенези мозолистого тела, его каудальный отдел (комиссура и тело) отсутствует в разной степени. Полная агенезия обычно считается мальформацией, возникающей в результате нарушения эмбриогенеза, в то время как частичная может представлять собой как истинную мальформацию, так и дисрупцию, которая произошла на каком-либо сроке беременности.
Кроме того, эхографические признаки частичной агенезии еще более трудно обнаруживаются, чем при наличии полной формы. В связи с этим антенатальная диагностика такого состояния во многих случаях бывает невозможна.
Прогноз при изолированной форме агенезии мозолистого тела остается неизученным. Многие авторы полагают, что агенезия мозолистого тела не приводит к значительным последствиям для неврологического развития. Однако величина показателя специфического риска в настоящее время неизвестна.
До настоящего времении получены данные только о 30 детях с пренатально установленным диагнозом изолированной агенезии мозолистого тела (у которых отсутствовали другие аномалии и был определен нормальный кариотип), длительность постантального наблюдения за которыми варьировала от нескольких месяцев до 11 лет. Нормальное или с пограничными нарушениями неврологическое развитие было отмечено в 26 случаях (87%).
Мозоль — это локальное утолщение поверхностных слоев кожи, возникающее под воздействием давления или трения чаще всего на кистях и ступнях. Различают сухие и мокрые мозоли. Последние выглядят как обычные ранки и требуют соответствующего лечения. Сухие мозоли представляют собой плотные ограниченные участки омертвевшей кожи. Регулярная обработка сухой мозоли при проведении педикюра или маникюра и ношение специальных защитных средств, как правило, приводит к ее постепенному уменьшению и исчезновению. Лишь в некоторых случаях необходимо хирургическое лечение мозоли.
МКБ-10

Общие сведения
Обычно дерматология сталкивается с проблемой мозоли при развитии осложнений в виде инфицирования или в случаях, когда длительные попытки пациента избавиться от мозоли не привели к желаемым результатам. Большинство мозолей проходят при регулярном удалении слоев огрубевшей кожи с их поверхности. Эту процедуру обычно проводят в кабинетах маникюра и педикюра.
Причины мозоли
При постоянном трении или давлении на определенном участке кожи происходит интенсивное омертвение ее поверхностных слоев. При этом отмершие клетки эпидермиса не успевают отслаиваться и удаляться с поверхности кожи. В результате происходит наслоение и уплотнение омертвевшего эпидермиса с образованием мозоли. При повышенной потливости пот размягчает омертвевшую кожу и образуется мягкая мозоль. Под слоем утолщенного эпидермиса может скапливаться лимфа, что приводит к появлению водяной мозоли.
Образование мозоли на кистях связано с постоянным трением кожи рук о рукоятку инструмента (молоток, стамеска), садового инвентаря (тяпка, секатор), спортивного снаряда (перекладина, брусья, теннисная ракетка) или музыкального инструмента (у скрипачей, гитаристов и др.). Мозоль может возникать на коже локтей и коленей.
На ступнях мозоль чаще всего возникает из-за неудобной и слишком узкой обуви. Так туфли на высоком каблуке вызывают сдавление переднего отдела стопы, слишком тонкая подошва увеличивает нагрузку на свод стопы, а грубые внутренние швы обуви могут оказывать дополнительное трение. Постоянное трение определенных участков кожи стопы с образованием мозоли может быть обусловлено неправильной походкой или занятием спортом (бегуны, лыжники). Часто мозоли бывают связаны с различными заболеваниями и деформациями стопы: бурсит или артрит суставов стопы, деформирующий остеоартроз, плоскостопие, молоткообразные пальцы стопы, пяточная шпора.
Симптомы мозоли
Твердая мозоль представляет собой плотное ограниченное утолщение кожи желтоватого или желто-серого цвета немного возвышающееся над поверхностью кожного покрова. Твердая мозоль отличается малой чувствительностью и сама по себе обычно не вызывает боли. Болевой синдром обусловлен надавливанием на мозоль при работе с инструментом или ходьбе, при сдавлении тесной обувью.
Мягкая мозоль имеет вид открытой ранки и характеризуется выраженной болезненностью. Водяная мозоль или водянка представляет собой содержащий серозную жидкость пузырь. При вскрытии пузыря образуется ранка, прикрытая сверху остатками омертвевшей кожи пузыря.
Отдельно выделяют стержневую мозоль. Чаще всего такие мозоли образуются на коже подушечек в области III и IV межпальцевых промежутков. Они могут встречаться на тыльной стороне пальцев и изредка на подошве. Стержневая мозоль — это сухая мозоль, имеющая глубоко уходящий под кожу корень. В центре мозоли имеется отверстие, из которого видна его «шляпка». Обычно образование стержневой мозоли происходит на месте проникновения под кожу мелкого камешка или занозы.
Осложнения
При неправильном лечении мозоли, ее срезании, образовании на ее поверхности трещин может произойти инфицирование. Проникающие при этом в мозоль микроорганизмы вызывают ее воспаление, что проявляется покраснением, отеком и выраженной болезненностью мозоли. Без должного лечения происходит размягчение мозоли и при надавливании на нее начинает выделяться гной. Воспалительный процесс с мозоли может распространяться на окружающие ткани с развитием мозольного абсцесса или флегмоны, на кости стопы с возникновением остеомиелита, на синовиальные оболочки и суставы стопы.
Диагностика мозоли
Диагностировать мозоль по характерному внешнему виду совсем не трудно. Необходимо отличать мозоль от заусенец, воспалительных изменений в суставах плюсневых костей, болезни Мортона, генетически обусловленного повышенного ороговения кожи. Некоторые бородавки, особенно подошвенные, по внешнему виду могут напоминать мозоль. Отличительным признаком является большая чувствительность бородавки и возникновение болезненности при ее прокручивании, в то время как мозоль болит при надавливании.
Если пациент обратился на консультацию дерматолога, то врач обязательно должен выяснить, с чем связано образование мозоли. Для этого он будет расспрашивать пациента про его работу, хобби, спортивные и другие увлечения, характер обуви, которую он обычно носит. При локализации мозоли на стопе врач проведет осмотр, направленный на выявление деформаций и заболеваний стопы, при необходимости направит пациента к подологу, ревматологу или ортопеду. Важное значение имеет наличие у пациента таких заболеваний как сахарный диабет, неврит, облитерирующий эндартериит, варикозная болезнь с хронической венозной недостаточностью. При выявлении в анамнезе таких заболеваний для определения наиболее адекватной тактики лечения мозоли требуется консультация соответственно эндокринолога, невролога, сосудистого хирурга или флеболога.
Лечение мозоли
Как правило, не сопровождающаяся болевым синдромом мозоль не требует лечения. При возникновении боли необходимо устранить фактор ее вызывающий: узкую обувь, трение о рукоятку инструмента и т. п. Смягчить трение и давление на мозоль, возникающие при ходьбе, помогают специальные защитные подкладки для мозоли. Такую подкладку можно сделать самостоятельно. Из мягкой и достаточно толстой ткани вырезают круг с отверстием в центре. Круг прикладывают так, чтобы в центральном отверстии была мозоль. Если мозоли находятся под пальцами ног, используют специальную плюсневую подкладку, изготовленную из войлока, резины или мягкого пластика. При расположении мозоли на пальцах применяют прокладоки между пальцами, рукава и чехлы для пальцев. Разделяющие пальцы прокладки устраняют их трение друг о друга. Рукава и чехлы, которые одеваются на пальцы, защищают их боковую поверхность и кончики.
В случае сухой мозоли обработку мозоли проводят в салонах красоты как отдельную процедуру или в ходе выполнения педикюра или маникюра. Для этого применяют специальные размягчающие мозоль составы, салициловую кислоту, аппаратный педикюр. Мягкие мозоли и вскрывшиеся водянки обязательно обрабатывают антисептиками с наложением защитной повязки, которую фиксируют пластырем.
При выявлении деформаций стопы по возможности проводится ортопедическое лечение. В случае сопутствующей патологии, приводящей к нарушению иннервации или кровоснабжения в области мозоли, лечение должно проводиться совместно с соответствующим специалистом. Если консервативные методы лечения мозоли не приносят результата, необходимо ее удаление. Может быть проведена криодеструкция мозоли, ее удаление лазером, электрокоагуляция, удаление радиоволновым методом или хирургическое иссечение.
Профилактика образования мозоли
Профилактические мероприятия, направленные на предупреждение образования мозоли, состоят в ношении мягкой, достаточно свободной обуви с хорошей подошвой; применении защитных перчаток при работе с инструментами, наколенников и других защитных средств на места, подвергающиеся постоянному трению. Правильный уход за кожей кистей и стоп, регулярное применение смягчающих кремов, обработка пемзой участков повышенного ороговения кожи также способствует предупреждению образования мозолей.
Агенезия мозолистого тела — это врожденное отсутствие мозолистого тела либо его части. Аномалия обусловлена генетическими нарушениями, сосудистыми мальформациями, тератогенными факторами. Основные признаки заболевания: двигательные расстройства, задержка психоречевого развития, судорожные приступы. При негрубом (частичном) варианте патологии возможно малосимптомное течение. Для диагностики состояния назначается церебральные КТ или МРТ, нейросонография у новорожденных, генетические исследования. Лечение симптоматическое: медикаментозная коррекция осложнений, реабилитационные программы.
МКБ-10
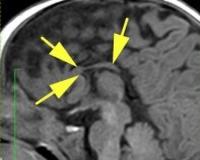
Общие сведения
Агенезия мозолистого тела (АМТ) — один из наиболее частых пороков нервной системы. Распространенность болезни в популяции составляет от 0,05% до 7% среди новорожденных, причем в группе детей с замедленным становлением психики агенезия встречается у 2,3%. Калифорнийская программа по изучению врожденных пороков предоставляет другие данные по частоте агенезии — 1,4 на 10000 живых новорожденных. Впервые состояние было описано в 1812 году в ходе аутопсии, проведенной немецким анатомом И. Рэйлем, и названо «природной моделью рассеченного мозга».
Причины
Точные этиологические факторы заболевания не установлены. В современной неврологии преобладает мультифакториальная теория, согласно которой для формирования врожденного порока ЦНС требуется комбинация неблагоприятных экзогенных и эндогенных причин. Ученые выделяют несколько наиболее вероятных предпосылок развития агенезии:
- Генетические аномалии. Повреждения мозолистого тела отмечаются при различных наследственных синдромах: Миллер-Дикера, Рубинштейна-Тауби, Доннаи-Кугана. Состояние входит в состав не менее 7 аутосомно-доминантных, 23 аутосомно-рецессивных, 12 Х-сцепленных врожденных заболеваний.
- Сосудистые нарушения. Причиной недоразвития мозолистого тела могут выступать артериовенозные мальформации или аневризмы, которые характеризуются отсутствием нормальной капиллярной сети. При этом возникает феномен обкрадывания, клетки МТ не получают должного количества кислорода, питательных веществ.
- Токсические влияния. Болезнь связана с действием тератогенных химических факторов: лекарственных препаратов, солей тяжелых металлов, пестицидов и бытовой химии. Негативное влияние на формирование ЦНС плода оказывает вдыхание табачного дыма (активное или пассивное курение) или прием беременной алкоголя во время гестации.
- Внутриутробные инфекции. Аномалии формирования неврологических структур, в том числе агенезия мозолистого тела, встречаются при проникновении возбудителей в организм плода на 2-3 месяце беременности. Нейротропные свойства демонстрируют герпетические инфекции, токсоплазмоз, цитомегаловирус.
Основным фактором риска выступает недоношенность. У новорожденных, родившихся до 27-недельного срока гестации МТ истончено в задних отделах, между 28 и 30 неделями — только в области валика. У рожденных после 30 недели в неонатальном периоде изменения не обнаруживаются, хотя при нейропсихологическом исследовании у школьников зачастую выявляется дефицит межполушарной передачи познавательной информации.
Патогенез
Агенезия возникает при нарушении дифференциации нервной трубки в период со 2 до 5 месяца внутриутробного развития. При полном отсутствии МТ третий мозговой желудочек остается открытым, не формируются столбы свода мозга, отсутствуют прозрачные перегородки. В 60% случаев при АМТ передней комиссуры нет вообще. В 10% она увеличена и берет на себя часть функций мозолистого тела у новорожденных, а также на следующих этапах постнатального периода.
Характерным анатомическим изменением является колпоцефалия, при которой расширены задние отделы боковых церебральных желудочков. Состояние не относится к истинной гидроцефалии новорожденных, а обусловлено уменьшением кортикальных ассоциативных путей. Еще один типичный признак порока — пучки Пробста, представляющие собой неправильно ориентированные аксоны, расположенные параллельно межполушарной щели.
Классификация
В практической неврологии состояние подразделяют на тотальное, когда орган полностью отсутствует, и частичное (парциальное), при котором визуализационные методы не обнаруживают отдельные участки МТ. Это имеет решающее значение для тяжести клинической картины, возможных осложнений. В соответствии с патогенетическими особенностями формирования врожденных пороков, выделяют следующие 3 формы болезни:
- Агенезия. Закладка эмбрионального зачатка МТ отсутствует полностью.
- Аплазия. Эмбриональный зачаток мозолистого тела есть, но не развивается.
- Гипоплазия. МТ недостаточно развито из-за нарушений на одном из этапов эмбриогенеза: размеры и масса органа уменьшены, его функциональная активность снижена.
Симптомы
Клиническая картина агенезии мозолистого тела широко варьирует от практически бессимптомных форм (при гипоплазии) до критических нервно-психических расстройств при его грубом недоразвитии, сопровождающемся другими врожденными пороками ЦНС. У новорожденных признаки патологии могут вовсе отсутствовать и проявляться по мере взросления младенца задержкой психомоторного развития.
Двигательные нарушения определяются у 35-40% пациентов. Они проявляются мышечной гипотонией или дистонией, гипер- или гипорефлексией, нарушением глотательного и сосательного рефлексов. Дети позже начинают держать голову, испытывают затруднения при обучении сидению, ползанию, ходьбе. Могут отмечаться координационные нарушения, неуклюжая походка. Из пароксизмальных расстройств у новорожденных и детей первого года жизни преобладают судороги.
Мозолистое тело поддерживает связь между церебральными зонами, формирует межполушарную организацию высших психических процессов. При его агенезии либо гипоплазии у детей выявляются когнитивные расстройства. У новорожденных пациентов и в раннем детстве наблюдается задержка речи, снижение динамического компонента игровой деятельности. В дошкольном и школьном возрасте возникают проблемы с концентрацией внимания, расстройства памяти, при тотальной АМТ снижен коэффициент интеллекта.
Осложнения
Около 65% случаев заболевания сопровождаются сопутствующими врожденными патологиями, среди которых преобладают мальформации кортикального развития (22,8%), межполушарные кисты (14,3%), голопрозэнцефалия (14,3%). К более редким сопутствующим аномалиям относят кисты и гипоплазию мозжечка, синдром Арнольда-Киари. До 20% новорожденных, кроме структур ЦНС, имеют пороки нескольких внутренних органов.
У 75% больных с тотальным поражением наблюдается симптоматическая эпилепсия височно-лобной локализации, в 66% случаев выражены когнитивные нарушения. У 16% пациентов формируются расстройства аутистического спектра. Изредка встречаются патологии органа зрения в виде хориоретинальных лакунарных очагов, сочетанной аномалии зрительных нервов.
Диагностика
В качестве первичного метода обследования в пренатальном периоде проводится акушерское УЗИ. У новорожденных для скрининговой диагностики используется нейросонография, однако этот метод не всегда показывает хорошую информативность, особенно при парциальной агенезии. Для верификации диагноза назначаются следующие методы исследования:
- КТ головного мозга. При компьютерной томографии определяются широко расставленные передние рога, высокое стояние третьего желудочка, параллельный ход медиальных стенок боковых желудочков. КТ производится в рамках постнатальной диагностики.
- МРТ головного мозга. Для максимально точной визуализации степени агенезии или гипоплазии мозолистого тела новорожденным выполняется магнитно-резонансная томография в трех плоскостях. По показаниям МРТ может рекомендоваться беременным женщинам для исключения несовместимых с жизнью сочетанных пороков ЦНС.
- Нейропсихологическое обследование. Для изучения когнитивных функций у детей применяется шкала интеллекта Векслера (WISC-Revised), адаптированное чтение и правописание (Schonnel Graded Reading and Spelling Tests), оценка вербальной беглости, тест контролируемых устных ассоциаций (Controlled Oral Word Association Test).
- Генетический анализ. Для подтверждения или исключения наследственных заболеваний, сопровождающихся агенезией мозолистого тела, показаны кариотипирование, секвенирование генома, проводимое как новорожденным, так и детям другого возраста. Исследования также проводят в антенатальном периоде для принятия решения о сохранении или прерывании беременности.
Лечение агенезии мозолистого тела
Специфическая терапия отсутствует. Медикаментозное лечение назначается неонатологом или педиатром индивидуально с учетом ведущих патологических синдромов: у новорожденных, детей раннего возраста используются антиконвульсанты, нейрометаболические препараты, дегидратационная терапия. Основу медицинской помощи составляет комплексная реабилитация, которая включает следующие составляющие:
- Нейрологопедические программы. Занятия с детским логопедом проводятся для становления речевой функции, ликвидации проявлений дизартрии, улучшения артикуляции.
- Дефектологические программы. Помощь коррекционных педагогов требуется детям с интеллектуальными нарушениями, которые не могут проходить обучение в обычных классах.
- Нейроакустические программы. Формирование и гармонизация высших психических функций производятся с помощью звуковой терапии, музыкотерапии.
Прогноз и профилактика
Прогноз определяется видом врожденной аномалии мозолистого тела, наличием сопутствующих пороков развития ЦНС. Благоприятный исход наблюдается при частичной гипоплазии МТ, а в случае комбинированных церебральных пороков у новорожденных могут быть жизнеугрожающие осложнения. Профилактические меры включают медико-генетическое консультирование, исключение тератогенных влияний в гестационном периоде.
1. Эпилептические проявления когнитивные и аутистические расстройства у пациентов с агенезией мозолистого тела: результаты нейропсихологического тестирования/ О.А. Милованова, О.А. Комиссарова, Т.Ю. Тараканова, С.В. Бугрий// Эпилепсия и пароксизмальные состояния. — 2018. — №4.
2. Влияние особенностей строения мозолистого тела и доминирующего полушария на протекание психических процессов в юношеском возрасте/ У.С. Чернышова, Т.Ю. Хабарова, Д.А. Соколов// Центральный научный вестник. — 2016.
4. Молекулярная эмбриология: на пути каталогизации генов врожденных пороков развития головного мозга/ В.П. Пишак, М.А. Ризничук// Международный журнал педиатрии, акушерства и гинекологии. — 2014. — №5.
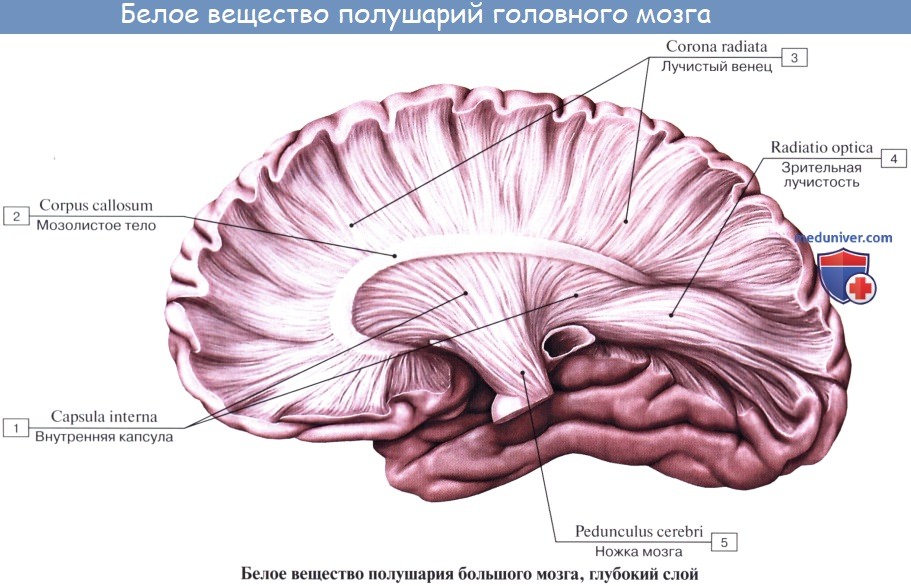
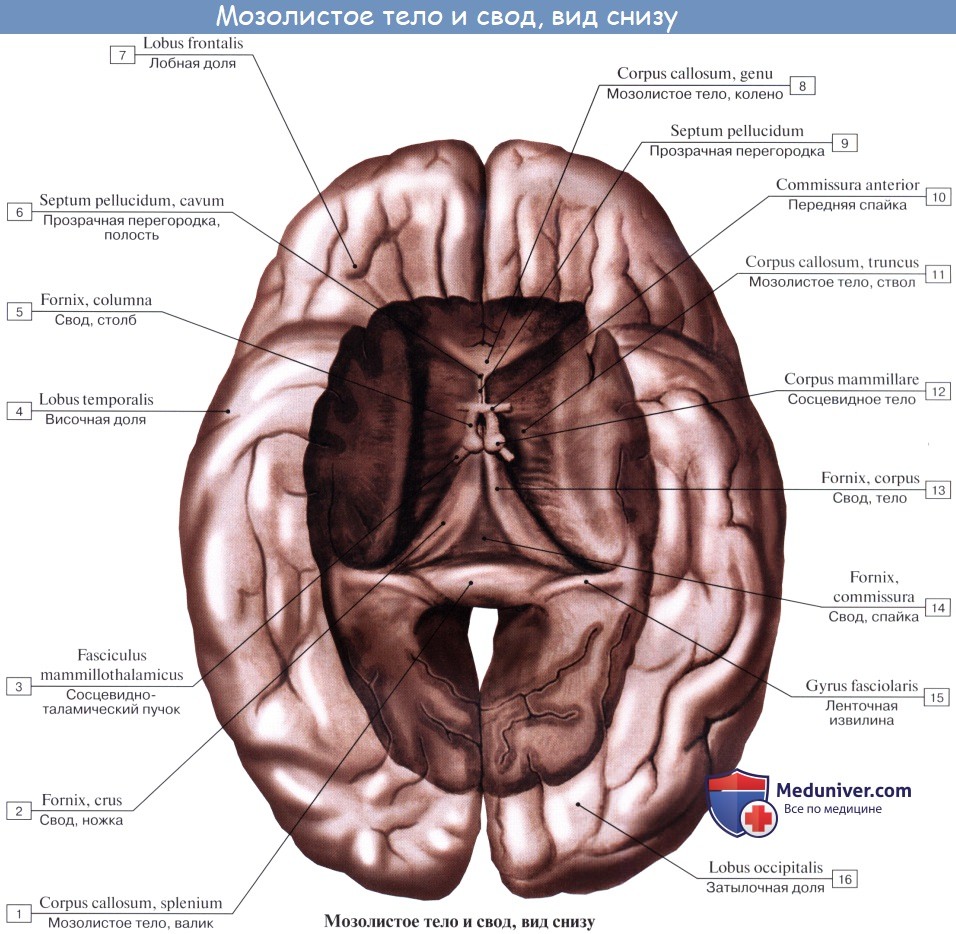
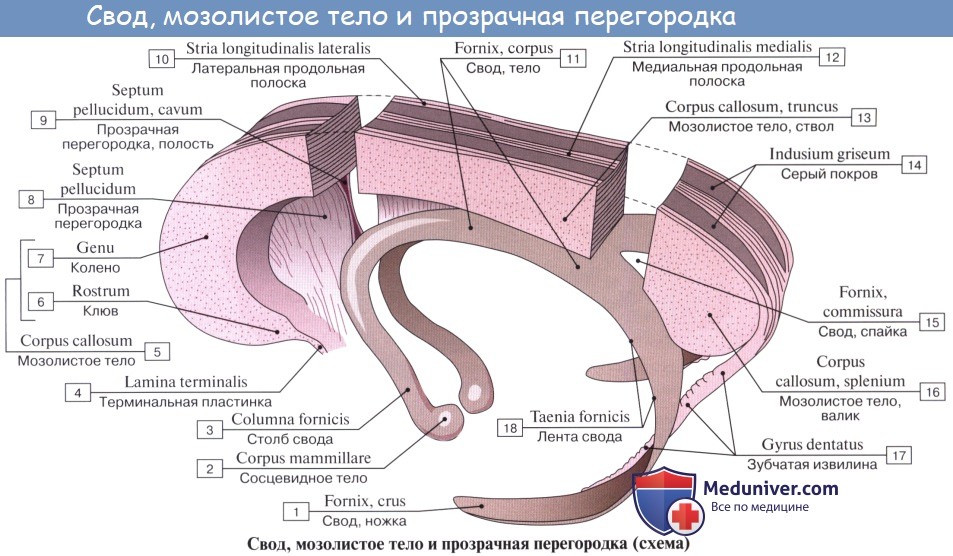
Мозолистое тело, corpus callosum. Колено мозолистого тела
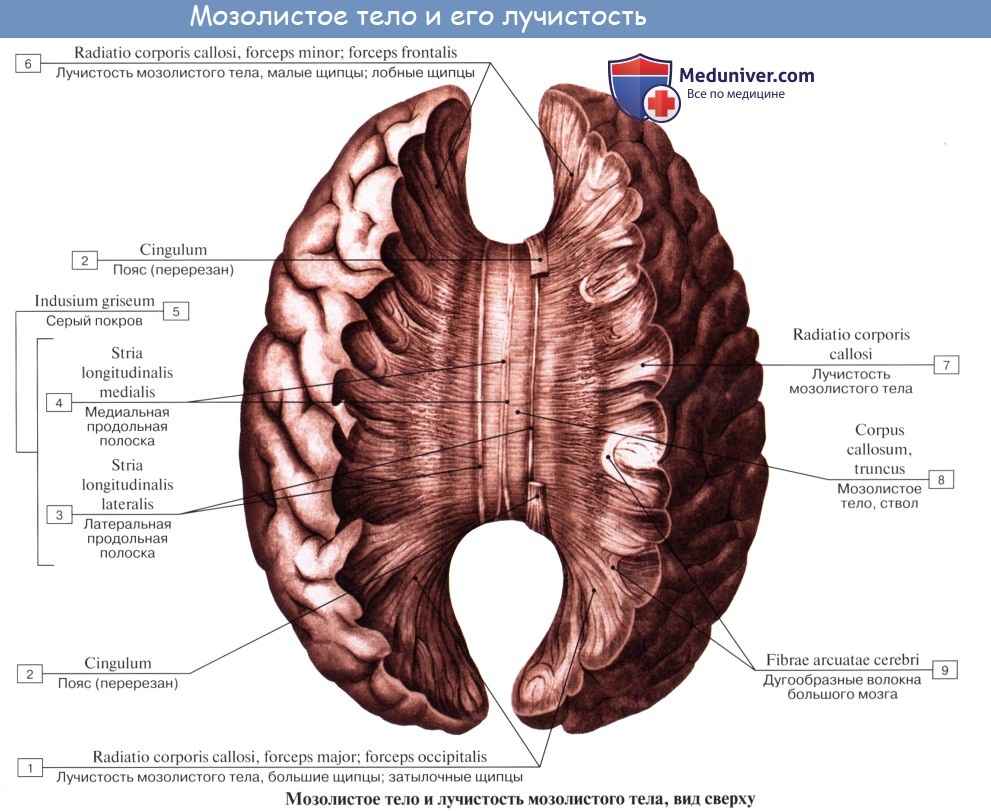
В глубине продольной щели мозга оба полушария соединены между собой толстой горизонтальной пластинкой — мозолистым телом, corpus callosum, которое состоит из нервных волокон, идущих поперечно из одного полушария в другое. В мозолистом теле различают передний загибающийся книзу конец, или колено, genu corporis callosi, среднюю часть, тело, truncus corporis callosi, и затем задний конец, утолщенный в форме валика, splenium corporis callosi.
Все эти части хорошо видны на сагиттальном разрезе мозга между обоими полушариями. Колено мозолистого тела, загибаясь книзу, заостряется и образует клюв, rostrum corporis callosi, который переходит в тонкую пластинку, lamina rostralis, продолжающуюся в свою очередь в lamina terminalis.
Видео урок для зубрешки анатомия базальных ядер, внутренней капсулы, белого вещества и волокон полушарий мозга
Редактор: Искандер Милевски. Дата последнего обновления публикации: 13.8.2020
Информация на сайте подлежит консультации лечащим врачом и не заменяет очной консультации с ним.
См. подробнее в пользовательском соглашении.
Агенезия и дисгенезия мозолистого тела - клиника, диагностика
Агенезия и дисгенезия мозолистого тела Может быть полной или частичной. В последних случаях задняя часть обычно утрачена, из-за того, что мозолистое тело развивается в переднезаднем направлении, тем не менее, возможна и передняя агенезия (Aicardi et al., 1987; Barkovich и Norman, 1988b; Sztriha, 2005). Встречаются атипические формы, сложно отличимые от голопрозэнцефалии (Barkovich, 1990). Агенезия мозолистого тела относительно распространена. Распространенность среди населения в целом оценивается как 3-7/1000 (Bedeschi et al., 2006).
Наблюдаемая частота возросла с введением в практику КТ и MPT. Jeret et al. (1987) выявили 33 случая в серии 1447 КТ снимков.
Даже теперь диагноз в основном ставится только пациентам с неврологическими симптомами, поэтому истинная частота не известна.
Отсутствие мозолистой спайки, как правило, замещается двумя продольными связками, известными как продольное мозолистое тело или пучки Пробста, проходящие по внутренней стороне полушарий. Часто появляются борозды на внутренней стороне с радиальным расположением и расширением затылочных рогов, сохраняющих их фетальную морфологию, так называемую кольпоцефалию (Noorani et al., 1988). Другие связанные аномалии часто включают в себя формирование кист кзади от третьего желудочка, сообщающиеся или несообщающиеся (Yokota et al., 1984, Griebel et al., 1995, Barkovich et al., 2001b) аномалии червя мозжечка и ствола мозга, а также смешанные мальформации ЦНС, такие как гетеротопия, аномалии образования извилин или цефалоцеле (Jeret et al., 1987; Barkovich и Norman, 1988b; Serur et al., 1988).
Гигантские кисты могут иметь благоприятный исход, несмотря на внушительные размеры (Lena et al., 1995; Haverkamp et al, 2002). Пороки развития ЦНС были обнаружены у 33% пациентов с полной и у 42% из них с частичной агенезией (Bedeschi et al., 2006).
Именно эти сочетанные аномалии отвечают за клинические проявления. Липома мозолистого тела почти неизменно сопровождает агенезию этой структуры (Zee et al., 1981; Vade и Horowitz, 1992). Характерны периферические мальформации (Parrish et al., 1979). Особенно часто встречаются глазные аномалии (Aicardi et al., 1987). Гипоплазия мозолистого тела (Bodensteiner et al., 1994) может быть минимальной формой каллозной дисгенезии, но гораздо чаще следствием кортикальных нейрональных потерь.
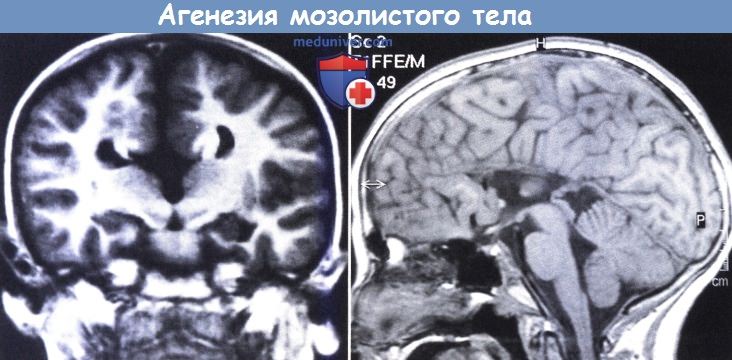
Агенезия мозолистого тела, (слева) МРТ (инверсия-восстановление):
продольное мозолистое тело (пучки Пробста) около внутренней поверхности тел желудочков мозга.
(справа) Сагиттальная проекция: полное отсутствие мозолистого тела и радиальное распределение борозд на внутренней стороне полушарий.
Serur et al. (1988) просмотрели 81 случай из литературных источников, из которых в 21 была трисомия 8, у 14 была трисомия 13-15 и у 18 затронуты хромосомы 17 или 18. Из 34 выполненных кариотипов в двух была обнаружена трисомия 8. Субтеломерные абберации были выявлены в 5% случаев Bedeschi et al. (2006). Среди экологических факторов выделяют плодный алкогольный синдром. Некоторые метаболические заболевания, особенно гипергликемия (Dobyns, 1989), недостаточность пируватдегидрогеназы (Bamforth et al., 1988, Raoul et al., 2003) и другие метаболические расстройства вместе составляют около 2% случаев агенезии мозолистого тела. В большинстве случаев их происхождение неизвестно.
Описание клинических проявлений каллозной аге-незии можно разделить на две части: несиндромные и синдромные формы (Davila-Guttierez, 2002).
Несиндромные формы наиболее распространены (Jeret et al., 1987; Serur et al., 1988). Неизвестный процент случаев остается бессимптомным или случайно выявляется только благодаря большим размерам головы. У большинства пациентов отмечается задержка умственного развития, судороги и/или большие размеры головы (Aicardi et al., 1987). Часто обнаруживаются гипертелоризм. В исследовании Jeret et al. (1987) 82% пациентов имели умственную отсталость или задержку развития, 43% страдали судорогами и у 31% развился церебральный паралич.
Однако нормальное когнитивное развитие наблюдалось у 9 из 63 детей (Bedeschi et al., 2006), возможно, и чаще, поскольку бессимптомные случаи, вероятно, не диагностированы. Возможны судороги любого типа, включая инфантильные спазмы, но чаще — очаговые. Хотя характерно увеличение размеров головы, иногда более 5-7 СО от среднего, показания к шунтированию достаточно строги, так как многие случаи «гидроцефалии» спонтанно стабилизируются, не причиняя каких-либо проблем. Макроцефалия может быть частично связана с наличием гигантских кист, расположенных кзади от третьего желудочка (Barkovich et al., 2001b).
Специфические расстройства межполушарной передачи либо отсутствуют, либо только минимальные (Jeeves и Temple, 1987).
Синдромные формы перечислены в таблице ниже.
Диагноз определяют хориоидальные лакуны и сопутствующие аномалии, выявленные при МРТ (перивентрикулярная гетеротопия, диспластичная кора, эпендимальные кисты). При патологическом исследовании в мозге обнаруживают многочисленные участки гетеротопии и полимикрогирии не разделенного на слои типа (Billette de Villemeur et al., 1992), тогда как так называемые лакуны представляют собой истончение пигментного эпителиального и сосудистого слоя с утратой пигментных гранул. Эпендимальные кисты часто обнаруживают вокруг третьего желудочка. Кисты или опухоли сосудистого сплетения могут достигать больших размеров (Aicardi, 2005). При выявлении вместе с агенезией мозолистого тела возможен пренатальный диагноз.
Другие синдромные формы встречаются редко или в большинстве ограничены определенными этническими группами.
Синдром Айкарди у трехмесячной девочки.
Обратите внимание на асимметрию полушарий, двусторонние кисты сосудистого сплетения, кисты вокруг третьего желудочка с различным сигналом от цереброспинальной жидкости и перивентрикулярные гетеротопии.
Эпендимальное происхождение кист сосудистого сплетения было подтверждено при гистологическом исследовании. Агенезия мозолистого тепа. Ультразвуковое антенатальное исследование, сагиттальная проекция.
Обратите внимание на нормальный четвертый желудочек, отсутствие эхосигнала от мозолистого тела и расширенный боковой желудочек.
Слева на фотографии — затылок плода.
Семейный синдром агенезии мозолистого тела с патологией гениталий, который также может проявляться с микроцефалией и другими аномалиями ЦНС, является частью более обширного спектра расстройств, связанных с мутациями в гене ARX на хромосоме Хр22.3 (Hartmann et al, 2004).
Синдром Андерманна был описан у французского канадца в районе озера Сент-Джонс (Andermann, 1981), но о нескольких случаях было заявлено за пределами Канады. Этот синдром затрагивает периферическую нервную систему в дополнение к агенезии мозолистого тела или гипотрофии. Агенезия мозолистого тела зачастую является частью рото-лице-пальцевого синдрома I типа.
Диагноз агенезии каллозного тела основывается на данных нейровизуализации. Диагноз полной агенезии несложен при ультрасонографии, КТ и MPT (Aicardi et al., 1987; Jeret et al., 1987; Serur et al., 1988). MPT эффективнее при диагностике частичной агенезии. Диагностика методами нейровизуализации не представляет сложностей, при КТ или МРТ выявляется подъем третьего желудочка и широкое разделение передних рогов с классической картиной «бычьих рогов» на фронтальных срезах. Диффузионно-тензорная МР (Lee et al., 2005) позволяет выявить отклонение трактов, особенно пучков Пробста, которые направляются кзади около стенки желудочка и не пересекают противоположную сторону.
Пренатальная диагностика возможна с 22 недели (Bennett et al., 1996; Simon et al., 2000a). Решение о прерывании беременности сложно принять безоговорочно, пока нет данных о распространенности бессимптомных случаев. Blum et al. (1990) сообщали, что у 6 из 12 новорожденных, у которых агенезия мозолистого тела была диагностирована антенатально, имели нормальное развитие в возрасте 2-8 лет. Moutard et al. (2003) наблюдали 17 случаев с пренатально диагностированной изолированной агенезией с повторными измерениями IQ. В возрасте 6 лет все дети имели коэффициент умственного развития на нормальном уровне с тенденцией к нижней границе нормы. Девять детей в исследовании Bedeschi et al. (2006) имели нормальное развитие.
Тем не менее, у 2 из 9 детей, пренатально диагностированных с помощью МРТ без сочетанных аномалий, отмечалась задержка в развитии (Volpe et al., 2006). Случаи, связанные с другими мальформациями или хромосомными аномалиями, неизменно имели неблагоприятный исход. Поэтому крайне важно фетальное кариотипирование и полное обследование на наличие сочетанных пороков развития.
Читайте также: