
Врожденный буллезный эпидермолиз дистрофическая форма что это такое
Обновлено: 03.05.2024
ГБУЗ «Московский научно-практический центр дерматовенерологии и косметологии» Департамента здравоохранения Москвы, Москва, Россия
Московская медицинская академия им. И.М. Сеченова
ГОУ Оренбургская государственная медицинская академия
ГБУЗ «Московский научно-практический центр дерматовенерологии и косметологии Департамента здравоохранения Москвы», Москва, Россия, 119071
Клинико-эпидемиологические особенности врожденного буллезного эпидермолиза в Москве
Журнал: Клиническая дерматология и венерология. 2017;16(6): 83‑89
ГБУЗ «Московский научно-практический центр дерматовенерологии и косметологии» Департамента здравоохранения Москвы, Москва, Россия
Врожденный буллезный эпидермолиз (далее — ВБЭ) является тяжелым врожденным заболеванием. В Москве, как в мегаполисе, находится большое количество больных с данным диагнозом. В настоящее время во всем мире проводят передовые исследования, направленные на разработку генных, клеточных и белково-заместительных методов терапии больных ВБЭ. Тем не менее больные ВБЭ нуждаются в ежедневной помощи в виде средств наружной терапии и перевязочных материалов. Цель исследования — проанализировать распространенность ВБЭ и изучить клинические особенности различных форм ВБЭ в Москве. Материал и методы. Материалом послужили данные Московского научно-практического центра дерматовенерологии и косметологии Департамента здравоохранения Москвы за 2013—2017 гг. Результаты. Впервые установлена распространенность и структура заболеваемости ВБЭ в Москве, в том числе структура инвалидности среди детского и взрослого населения Москвы с различными формами ВБЭ. Заключение. Несмотря на то что ВБЭ считается орфанным заболеванием, пациент с данным генодерматозом может обратиться к практикующему врачу любой специализации.
ГБУЗ «Московский научно-практический центр дерматовенерологии и косметологии» Департамента здравоохранения Москвы, Москва, Россия
Московская медицинская академия им. И.М. Сеченова
ГОУ Оренбургская государственная медицинская академия
ГБУЗ «Московский научно-практический центр дерматовенерологии и косметологии Департамента здравоохранения Москвы», Москва, Россия, 119071
Врожденный буллезный эпидермолиз (ВБЭ) относится к группе редких генетических невоспалительных заболеваний кожи, характеризующихся склонностью кожи и слизистых оболочек к развитию рецидивирующих пузырей или эрозий, преимущественно на местах незначительной механической травмы (трение, давление, прием твердой пищи).
Несмотря на то что диагноз ВБЭ имеет код Q и относится по МКБ-10 к классу XVII «Врожденные аномалии (пороки развития), деформации и хромосомные нарушения», большинство пациентов обращаются с данной патологией кожного процесса к врачам дерматовенерологам.
Учитывая разнообразие отдельных фенотипических форм ВБЭ, тяжесть заболевания, торпидность течения, низкий потенциал реабилитации, а также социальный аспект, каждый пациент с диагнозом ВБЭ нуждается в постоянном поддерживающем комплексном лечении и уходе для поддержания качества жизни.
Частота ВБЭ не зависит от расовой и этнической принадлежности и может проявляется в равной степени у обоих полов. Точная распространенность простого ВБЭ (ПБЭ) во всем мире до сих пор неизвестна, но, по данным разных источников [1], заболеваемость ПБЭ может варьировать в пределах 1:30 000—50 000. Локализованный тип БЭ является наиболее распространенной формой генодерматоза, так как он крайне редко приводит к летальному исходу больного и является формой ПБЭ, которая в наименьшей степени приводит к ухудшению качества жизни пациента [2]. Тем не менее проблема заболеваемости ВБЭ, также как и проблема совершенствования генетической диагностики в первые дни жизни ребенка, до сих пор несомненно актуальна в крупнейшем по численности населения субъекте Российской Федерации — Москве.
Материал и методы
Исследование проводилось в ГБУЗ «Московский научно-практический центр дерматовенерологии и косметологии» (МНПЦДК) — крупнейшей медицинской организации Москвы, оказывающей специализированную медицинскую помощь населению по профилю «Дерматовенерология». В период с 2013 по 2017 г. обследован 51 пациент с диагнозом врожденный буллезный эпидермолиз. Все пациенты либо проживали в Москве и состояли на диспансерном учете в филиалах МНПЦДК, либо это были иногородние граждане, впервые самостоятельно обратившиеся в МНПЦДК с целью дополнительного обследования. В рамках обследования все пациенты были консультированы главным внештатным специалистом дерматовенерологом и косметологом Департамента здравоохранения Москвы директором МНПЦДК проф. Н.Н. Потекаевым и медицинским генетиком с целью верификации диагноза.
Результаты
В филиалах и структурных подразделениях МНПЦДК были изучены данные о 51 пациенте с диагнозом ВБЭ. Все больные ВБЭ были включены в специальный реестр, который постоянно актуализируется. Возраст пациентов составлял от 0 до 27 лет. В рамках обследования все пациенты или их законные представители подписали добровольное информированное согласие на обработку персональных данных. В исследуемой группе преобладали дети в возрасте до 18 лет — 34 (66,7%). Группу взрослых в возрасте старше 18 лет составили 17 (33,3%) человек (рис. 1). Рис. 1. Распределение больных ВБЭ на группы детей и взрослых. При проведении анализа детской группы больных ВБЭ было установлено, что в настоящее время диагноз ВБЭ установлен 12 детям дошкольного возраста, что составляет 23,5% среди всей группы исследуемых с диагнозом ВБЭ и 35,3% среди пациентов до 18 лет, 22 несовершеннолетним в возрасте от 7 до 18 лет (43,1% среди всех пациентов с диагнозом ВБЭ и 64,7% среди всех пациентов до 18 лет). Половину группы дошкольного возраста составили дети в возрасте от 1 года до 3 лет — 6 человек. Аналогичное количество больных ВБЭ на момент исследования относились к возрастным группам от 9 до 11 лет и от 13 до 15 лет (рис. 2, 3). Рис. 3. Распределение больных ВБЭ по возрастным группам. Рис. 2. Распределение больных ВБЭ по возрастным группам.
На основании данных гистограммы и графика, представленных на рис. 2 и 3, можно сделать вывод, что наибольшее количество пациентов с ВБЭ приходится на возраст с 1 года до 3 лет, с 9 до 11 лет и с 13 до 15 лет. Данные показатели вероятнее всего связаны с периодами проведения профилактических медицинских осмотров. В соответствии с «Перечнем исследований при проведении медицинских осмотров несовершеннолетних» (далее — Перечень), утвержденный приказом Министерства Российской Федерации № 1346н от 21.12.12 «О порядке прохождения несовершеннолетними медицинских осмотров, в том числе при поступлении в образовательные учреждения и в период обучения в них», дети в возрастных группах от 1 года до 3 лет, с 9 до 11 лет и с 13 до 15 лет проходят обследования у врачей следующих специальностей: педиатр, детский стоматолог, невролог, детский хирург, офтальмолог, оториноларинголог, детский или подростковый психиатр, детский уролог-андролог, акушер-гинеколог. Важно отметить, что несовершеннолетние в возрасте 9—11 лет проходят дополнительное обследование у детского эндокринолога, травматолога-ортопеда, всем детям проводится электрокардиографическое исследование. Пациентам в возрасте от 13 до 15 лет, кроме обследований у упомянутых выше специалистов, проводятся дополнительные исследования в виде электрокардиографии; ультразвукового исследования органов брюшной полости, сердца, щитовидной железы, органов репродуктивной сферы; флюорографическое исследование.
Контроль организации и качества диспансеризации взрослого и детского населения осуществляют территориальные фонды ОМС и страховые медицинские организации.
Диспансеризация детского населения проводится на основании приказов Министерства здравоохранения Российской Федерации: Федерального закона № 323-ФЗ от 21.11.11 «Об основах охраны здоровья граждан в Российской Федерации» № 1346н от 21.12.12 «О порядке прохождения несовершеннолетними медицинских осмотров, в том числе при поступлении в образовательные учреждения и в период обучения в них», приказа № 72н от 15.02.13 «О проведении диспансеризации пребывающих в стационарных учреждениях детей-сирот и детей, находящихся в трудной жизненной ситуации» (вместе с «Порядком проведения диспансеризации пребывающих в стационарных учреждениях детей-сирот и детей, находящихся в трудной жизненной ситуации»), приказа № 216н от 11.04.13 «Об утверждении Порядка диспансеризации детей-сирот и детей, оставшихся без попечения родителей, в том числе усыновленных (удочеренных), принятых под опеку (попечительство), в приемную или патронатную семью».
При проведении анализа нозологической группы больных с ВБЭ было установлено, что 2 детей (3,9% из всех пациентов с ВБЭ, 5,9% из пациентов до 18 лет) находятся на индивидуальной форме обучения, что обусловлено тяжестью течения заболевания и длительными периодами ухудшения патологического кожного процесса. Этим детям выставлен и генетически подтвержден диагноз генерализованный рецессивный дистрофический БЭ (Аллопо—Сименса).

Из общего числа пациентов (n=51) большую часть составили мужчины (n=32; 62,8%) (рис. 4). Рис. 4. Гендерное распределение больных ВБЭ.

При детальном изучении индивидуального и семейного анамнеза установлено, что у 16 (31,4%) пациентов наследственность отягощена наличием ВБЭ в анамнезе хотя бы у одного из биологических родителей. Из общего числа пациентов 3 (5,9%) человека имели отягощенную наследственность в виде других заболеваний кожи и подкожной жировой клетчатки, в том числе пузырных дерматозов хотя бы у одного из биологических родителей. Наследственность не отягощена заболеваниями кожи и подкожной жировой клетчатки у 24 (47,0%) пациентов. У 8 (15,7%) пациентов подробный сбор анамнеза был невозможен в связи с отсутствием данных об одном из биологических родителей. Данные представлены на рис. 5. Рис. 5. Семейный анамнез у больных ВБЭ. В анамнезе ВБЭ по материнской линии прослеживается у 14 (27,5%) пациентов, по отцовской линии – у 1 (2%) и у 1 (2%) ВБЭ в анамнезе имелся и по материнской, и по отцовской линиям. В 3 (5,9%) семьях ВБЭ в анамнезе прослежен в трех поколениях, в 2 (3,9%) семьях — в двух поколениях. И в первом, и во втором случае ВБЭ наследовался по материнской линии. Из общего числа пациентов родных братьев или сестер с диагнозом ВБЭ имеют 5 (9,8%) человек.

Установленный диагноз с указанием группы и формы ВБЭ имели 23 (45,1%) пациента из 51. У 28 (54,9%) диагноз ВБЭ установлен без уточнения клинической разновидности и формы (рис. 6). Рис. 6. Встречаемость клинических форм у больных ВБЭ.
Диагноз простой буллезный эпидермолиз (Q81.0) был выставлен 34 (66,7%) лицам, среди которых простой буллезный эпидермолиз Вебера—Коккейна (локализованный БЭ кистей и стоп) (Weber—Cockayne) диагностирован у 8 (23,5%) человек, что составило 15,7% от числа всех пациентов с ВБЭ. Генерализованный простой буллезный эпидермолиз Кебнера (Köbner) — у 2 (5,9%) пациентов, или 3,9% из числа всех больных ВБЭ; простой герпетиформный буллезный эпидермолиз БЭ (Доулинг—Меара, Dowling—Meara) — у 1 (2,9%) пациента (или 2% из всех пациентов с ВБЭ).
Диагноз пограничный (переходный) буллезный эпидермолиз Херлитца (Q81.1) выставлен клинически и генетически верифицирован 2 (3,9%) детям женского пола в возрасте 14 и 2 лет.
Диагноз дистрофический буллезный эпидермолиз установлен у 15 (29,4%) пациентов, у 10 (66,7%), 19,6% из всех пациентов с ВБЭ) из которых выявлен генерализованный рецессивный дистрофический БЭ (Аллопо—Сименса).
С более чем 1500 мутациями в 17 генах, кодирующими структурные белки кожи и обеспечивающими связь между эпидермисом и дермой, ассоциировано развитие различных клинических форм ВБЭ. При наличии мутации в ассоциированном гене происходит прекращение или уменьшение синтеза их продуктов-белков, либо синтезируемые белки оказываются функционально или структурно неполноценными.
Для верификации диагноза четырем детям проводились дополнительные методы диагностики ВБЭ. В двух семьях использовалось такое генетическое исследование (ДНК-диагностика), как метод прямого автоматического секвенирования. С помощью данного метода были обнаружены мутации гена, кодирующего коллаген VII типа (COL7A1) — важнейший компонент формирования якорных фибрилл в дермоэпидермальном соединении, локализованный под плотной темной пластинкой базальной мембраны эпидермиса (sublamina densa). Мутации в данном гене, как правило, являются причиной дистрофических форм буллезного эпидермолиза. Также методом прямого автоматического секвенирования у 1 ребенка были обнаружены мутации в гене, кодирующим кератин 5 (KRT5), локализованный в кератиноцитах базального слоя эпидермиса, что приводит к возникновению простых форм базального буллезного эпидермолиза. Методом иммунофлюоресцентного антигенного картирования (ИАК) 1 ребенку верифицирован диагноз рецессивный дистрофический буллезный эпидермолиз (Аллопо—Сименса).
На основании результатов и данных обследований, проведенных врачами смежных специальностей, у 34 (66,7%) пациентов выявлена мультиорганность поражения и различные множественные сопутствующие соматические патологии органов и систем, приводящие в ряде случаев к инвалидизации пациента и, как следствие, к ухудшению качества жизни. Вместе с тем у 30 (88,2%) пациентов диагностированы более чем две сопутствующие патологии.
Из общего числа пациентов (n=51) инвалидами детства являются 19 (37,3%) детей (55,9% из группы исследуемых до 18 лет). Инвалидность I—III группы имеют 7 (41,2%) человек в возрасте старше 18 лет, что составляет 13,7% от всех обследованных больных ВБЭ.

Нарушения опорно-двигательного аппарата (ОДА) выявлены у 23 пациентов — более чем у половины (67,5%) из группы исследуемых, имеющих сопутствующие заболевания. На втором месте среди сопутствующих заболеваний находится группа функциональных нарушений желудочно-кишечного тракта. Диагноз, относящийся к данной группе заболеваний, был установлен 19 (55,9%) пациентам. Самым распространенным сопутствующим заболеванием является нарушение моторики кишечника и функциональный запор (диагностированы у 5 человек). Патологии, связанные с эндокринными нарушениями, выявлены у 15 (44,1%) человек; заболевания ЛОР-органов — у 12 (35,3%), нарушения работы головного мозга, центральной нервной системы — у 9 (26,5%), органа зрения — у 9 (26,5%), анемии различной этиологии — у 8 (23,5%), патология сердечно-сосудистой системы диагностирована — у 8 человек. Стоматологические заболевания (поражения полости рта) при проведении профилактических осмотров выявлены у 8 (23,5%) пациентов; заболевания мочеполовой системы — у 7 (20,6%). Совместно с диагнозом ВБЭ у 7 пациентов также диагностированы небуллезные дерматозы и заболевания придатков кожи, что составляет 20,6% в группе исследуемых. У 2 (5,9%) больных с ВБЭ диагностированы грыжа белой линии живота и пупочная грыжа. Распределение сопутствующих заболеваний у больных ВБЭ визуально отражено на рис. 7. Рис. 7. Сопутствующие заболевания у больных ВБЭ.

В соответствии с данными, полученными при обследовании пациентов с ВБЭ, было проведено картирование пациентов по округам Москвы. Согласно результатам проведенного анализа, в Центральном административном округе (ЦАО) проживает 1 пациент с диагнозом ВБЭ (2%), в Северном (САО) — 5 (9,8%), в Северо-Восточном (СВАО) — 5 (9,8%), в Восточном (ВАО) — 5 (9,8%), в Юго-Восточном (ЮВАО) — 8 (15,7%), в Южном (ЮАО) — 6 (11,8%), в Юго-Западном (ЮЗАО) — 10 (19,6%), в Западном (ЗАО) — 7 (13,7%), в Северо-Западном (СЗАО) — 1 (2%), в Зеленоградском административном округе (ЗелАО) — 1 (2%) (рис. 8). Рис. 8. Распределение больных ВБЭ по округам Москвы. Пациенты, приехавшие из других регионов Российской Федерации, составили 3,9% (n=2) .
Таким образом, можно сделать вывод, что в САО (23,6%) проживает в 2 раза меньше детей с диагнозом ВБЭ, чем в ЮАО (47,1%). В ЗАО (13,7%) проживает на 3,9% больше детей с диагнозом ВБЭ, чем в ВАО.
Сохранение и укрепление здоровья детей и подростков — актуальные проблемы здравоохранения, так как они составляют фундаментальную основу трудового потенциала страны и ее национальной безопасности [3—5].
При активном и своевременном выявлении больных ВБЭ не только врачами дерматовенерологами, но и врачами смежных специальностей, при мульдидисциплинарном подходе к обследованию и лечению данной группы пациентов ожидаемо значительное снижение частоты сопутствующих патологий и осложнений. Это связано не только с медицинскими, но и с социальными аспектами.
Заключение
Согласно результатам исследования, в Москве преобладают дети с диагнозом простой БЭ (45,1%). Среди группы исследуемых старше 18 лет также преобладают больные с диагнозом простой БЭ (21,6%).
При проведении статистического клинико-эпидемиологического анализа представленных данных и результатов собственных исследований больных ВБЭ, в том числе при пересчете показателей заболеваемости ВБЭ в Москве на 100 тыс. населения, в соответствии с данными Территориального органа Федеральной службы государственной статистики по Москве, были получены низкие показатели распространенности заболевания [6]. Полученные результаты демонстрируют, что показатель распространенности ВБЭ на территории Москвы составил 0,4 случая на 100 тыс. населения. В соответствии со ст. 44, п. 1. Федерального закона № 323-ФЗ от 21.11.11 (ред. от 25.06.12) «Об основах охраны здоровья граждан в Российской Федерации», редкими (орфанными) заболеваниями являются заболевания, которые имеют распространенность не более 10 случаев заболевания на 100 тыс. населения [7].
На основании вышеизложенного можно заключить, что ВБЭ соответствует статусу орфанного заболевания. Таким образом, больные ВБЭ нуждаются в разработке комплекса стандартов профилактических и лечебно-диагностических мероприятий, а также критериев включения при диагностировании данного генодерматоза при проведении медико-социальной экспертизы.
Буллезный эпидермолиз – группа наследственных заболеваний, которые характеризуются легкой ранимостью кожи, отсюда второе название этих патологий - «механобуллезная болезнь». Основным симптомом служит развитие на поверхности кожных покровов пузырей с серозным содержимым, после чего на их месте возникают долго незаживающие эрозии. Диагностика различных типов буллезного эпидермолиза осуществляется при помощи иммуногистологических и генетических методик, а также на основании данных осмотра пациента и изучения его наследственного анамнеза. Специфического лечения не существует, однако правильная и комплексная симптоматическая терапия может в ряде случаев значительно улучшать состояние больного.
МКБ-10
Общие сведения
Буллезный эпидермолиз – это гетерогенная группа наследственных заболеваний кожи, которые характеризуются образованием пузырей и эрозий в ответ на незначительное механическое воздействие. Впервые данный термин был использован в 1886 году немецким врачом-дерматологом Генрихом Кёбнером, дальнейшие исследования продемонстрировали, что существует множество разновидностей этой патологии. Генетические исследования буллезного эпидермолиза показали, что он может наследоваться как аутосомно-рецессивно, так и аутосомно-доминантно, с ним ассоциированы мутации более чем 10 генов. Существенные различия имеются и в клиническом течении разных типов этого заболевания, встречаемость колеблется в пределах 1:30000-1:1000000.
Патогенез нарушений при буллезном эпидермолизе долгое время оставался малоизученным. Прорыв в этом направлении произошел с внедрением в медицинскую практику электронной микроскопии, которая помогла визуализировать ультраструктуру пораженных тканей кожи. Следующий важный шаг в изучении буллезного эпидермолиза был совершен с открытием иммуногистологических исследований (иммунофлуоресценция). В настоящее время именно эти методики играют важнейшую роль в диагностике данных заболеваний, уступая по точности лишь генетическому анализу. Ввиду того, что методы изучения буллезного эпидермолиза постоянно совершенствовались, претерпевала изменения и классификация форм этой группы заболеваний.
Причины буллезного эпидермолиза
Этиология буллезного эпидермолиза неодинакова у разных типов заболевания, что в некоторых случаях достаточно сильно осложняет диагностику. Простой буллезный эпидермолиз обусловлен мутациями генов KRT5 и KRT14, однако, по данным врачей-генетиков, нарушением структуры этих генов объясняется только 75% случаев заболевания этого типа. При этом в кожных покровах, предположительно, нарушается равновесие в системе «ферменты-ингибиторы», и некоторые белки становятся объектом атаки. При простом буллезном эпидермолизе это могут быть протеины базальной мембраны (альфа6-бета4-интегрин) и белки десмосом базального слоя эпидермиса – десмоплакин, плакофиллин-1. В результате при механическом воздействии происходит выделение ферментов, которые разрушают указанные белки, тем самым провоцируя цитолиз и разрушение структуры эпидермиса, приводя к образованию пузырей.
Причиной развития другой формы патологии – пограничного буллезного эпидермолиза – являются мутации в генах LAMB3, LAMA3 и некоторых других. Большинство из этих мутации наследуется по аутосомно-рецессивному механизму, объектом атаки разбалансированной ферментной системы становятся такие протеины, как коллаген 17-го типа и ламинин-332. Эти белки участвуют в поддержании нормальной структуры нижних слоев эпидермиса, поэтому их повреждение приводит к характерным клиническим симптомам пограничного буллезного эпидермолиза. Помимо легкого образования пузырей и эрозий он характеризуется также повышенной ломкостью кожных покровов и более тяжелым течением.
Дистрофический тип буллезного эпидермолиза обусловлен мутациями в гене COL7A1, которые могут наследоваться как по аутосомно-доминантному, так и аутосомно-рецессивному механизмам. Белком-мишенью при этом выступает коллаген 7-го типа, который отвечает за стабильность структуры других соединительнотканных волокон кожи. Уменьшение количества этого протеина в тканях кожных покровов приводит к легкому развитию высыпаний, эрозий и пузырей, а также нередко сопровождается нарушениями других органов. В частности, дистрофический буллезный эпидермолиз часто приводит к развитию контрактуры суставов, поражение захватывает слизистые оболочки органов дыхательной и пищеварительной систем. На рубцах, которые остаются после заживления эрозий, нередко возникают злокачественные опухоли.
В целом, общий патогенез буллезного эпидермолиза можно свести к нарушению активности некоторых ферментов в тканях кожи. В результате этого разрушаются определенные ключевые структурные белки эпидермиса, дермы или базальной мембраны, что нарушает связи между клетками и приводит к образованию пузырей при механическом воздействии даже незначительной силы. Типы буллезного эпидермолиза отличаются один от другого локализацией пузырьков, видом мутации, что привела к этому заболеванию, и разновидностью белка, который стал объектом атаки ферментов.
Классификация буллезного эпидермолиза
В настоящий момент существуют десятки разновидностей буллезного эпидермолиза, которые достаточно трудно классифицировать в определенные группы. Проблема осложняется еще и тем, что почти за полтора века изучения данной патологии предпринимались неоднократные попытки разделить ее на определенные типы, используя самые современные на тот момент данные. В конечном итоге это привело к некоторой путанице, даже в научной литературе можно найти самые разнообразные варианты разделения буллезного эпидермолиза на разновидности. Наиболее современная классификация этого состояния в дерматологии включает в себя четыре типа заболевания, которые, в свою очередь, делятся на ряд подтипов:
- Простой буллезный эпидермолиз – имеет 12 подтипов, наиболее распространенными из которых являются синдромы Вебера-Коккейна, Кёбнера, Доулинга-Меары. Может наследоваться как аутосомно-доминантно, так и рецессивно, встречаемость составляет 1:100000. Простой буллезный эпидермолиз характеризуется образованием внутриэпидермальных или, реже, субэпидермальных пузырей, так как при этом заболевании поражаются белки эпидермиса.
- Пограничный буллезный эпидермолиз – делится на 2 подтипа, один из которых имеет еще 6 самостоятельных клинических форм. Наиболее тяжелой формой этого заболевания является подтип Херлитца, имеющий крайне высокую смертность. Встречаемость пограничного буллезного эпидермолиза составляет около 1:500000, образование пузырей при нем происходит на уровне светлой пластинки, что и дало ему название «пограничный».
- Дистрофический буллезный эпидермолиз – имеет два подтипа, которые делятся по механизму наследования этой патологии (доминантный и рецессивный подтипы). При этом встречаемость доминантного варианта несколько выше (3:1000000 против 1:500000 у рецессивной формы дистрофического буллезного эпидермолиза). Рецессивная разновидность также имеет несколько клинических форм, наиболее тяжелой из которых является подтип Аллопо-Сименса. При этом варианте заболевания у больных возникают глубокие эрозии, оставляющие после себя шрамы, возможны контрактуры суставов, поражение слизистых оболочек. Образование пузырей при этом происходит в сосочковом слое дермы, что и обуславливает появление шрамов и длительное заживление эрозий.
- Синдром Киндлера, или смешанный буллезный эпидермолиз, является одной из наиболее редких и малоизученных форм данной патологии. Особенностью, которая позволила выделить эту форму в отдельный тип, является образование пузырей во всех слоях кожи – эпидермисе, у светлой пластинке, в дерме. В настоящий момент определен только белок, выступающий в качестве мишени ферментов при смешанном буллезном эпидермолизе – киндлин-1.
Такой тип разделения всех клинических форм буллезного эпидермолиза является в настоящее время общепринятым. Но даже в пределах одного типа наблюдается большое разнообразие клинических симптомов заболевания, что осложняет диагностику и нередко влияет на прогноз патологии. Поэтому на сегодняшний день не прекращаются поиски более структурированной и приемлемой классификации буллезного эпидермолиза.
Симптомы буллезного эпидермолиза
Проявления буллезного эпидермолиза разных типов объединяет одно – развитие пузырей и эрозий в ответ на механическое воздействие на кожу. Различается лишь степень выраженности этих изменений, локализация, время существования и результаты заживления. При локализованной форме простого буллезного эпидермолиза (подтип Вебера-Коккейна) поражения располагаются только на определенном участке тела (руки, стопы). В младенческом возрасте возможна более широкая площадь появления пузырей, но с возрастом их выраженность уменьшается. Напротив, генерализованный подтип Доулинга-Меары характеризуется развитием мелких везикулярных высыпаний на значительной площади тела. Такой тип буллезного эпидермолиза возникает с самого раннего детства и может стать причиной смерти ребенка, итогом разрешения пузырьков может быть гиперкератоз, нарушения пигментации кожи, иногда возникает поражение слизистых.
Пограничная форма буллезного эпидермолиза протекает намного более тяжело, особенно так называемый летальный подтип Херлитца. При этом наблюдается повышенная ломкость кожных покровов, образование большого количества пузырьков, эрозий, на лице и спине часто возникают симметричные грануляции. Поражаются и слизистые оболочки рта, обнаруживается гипоплазия эмали и обусловленный ею тяжелый кариес. Столь тяжелое течение пограничного буллезного эпидермолиза часто становится причиной летального исхода в первые годы жизни. У выживших больных во взрослом возрасте формируются контрактуры суставов, поражение почек, потеря ногтей. Более легкая атрофическая форма пограничного буллезного эпидермолиза также характеризуется обширными высыпаниями, после разрешения которых формируются атрофические участки и рубцы. Также она часто приводит к дистрофии ногтей и рубцовой алопеции.
Дистрофический буллезный эпидермолиз практически всегда является генерализованным и поражает обширные участки тела. Доминантный вариант заболевания в целом отличается более доброкачественным течением, образование пузырей и их разрешение происходит медленно, однако большинство больных в конце концов теряют ногти на руках. После заживления эрозий на поверхности кожи формируются заметные рубцы. Рецессивный вариант дистрофического буллезного эпидермолиза, особенно его тяжелый генерализованный подтип, протекает намного тяжелее: помимо высыпаний у больных часто регистрируются псевдосиндактилии, обширные шрамы, потеря ногтей. Возникает поражение костей скелета, на месте заживших шрамов с годами может развиваться плоскоклеточный рак. Проблемой является еще и высокая устойчивость подтипа Аллопо-Сименса к терапевтическим мероприятиям.
Осложнения любого типа буллезного эпидермолиза сводятся к риску развития шока (при обширных поражениях), присоединения вторичной инфекции и спровоцированного ею сепсиса, обезвоживания больных. В большинстве случаев терапевтические процедуры производят только с целью недопущения этих состояний. Вероятность развития осложнений тем выше, чем большую область тела занимают патологические очаги и чем деструктивнее их характер (напряженные пузыри, эрозии, язвы).
Диагностика буллезного эпидермолиза
В настоящее время диагностика буллезного эпидермолиза осуществляется путем осмотра кожных покровов пациента, с помощью проведения иммуногистологических исследований и генетических анализов, в некоторых случаях производят изучение наследственного анамнеза. При осмотре кожных покровов специалист также может произвести диагностические тесты – механически воздействовать на кожу пациента и спустя время оценить результаты. Развитие на этом участке характерных для буллезного эпидермолиза пузырей или эрозий говорит в пользу наличия данного заболевания. На следующих этапах диагностики производят более точное определение формы патологии.
Иммунофлуоресцентный анализ при буллезном эпидермолизе осуществляется при помощи моно- и поликлональных антител, имеющих сродство к основным белкам эпидермиса, светлой пластинки и верхних слоев дермы. Это позволяет оценить количество того или иного белка, что, в свою очередь, говорит о ферментной активности тканей. Уменьшение количества того или иного белка свидетельствует о его низком выделении или же ускоренном разрушении. Снижение концентрации ключевых протеинов на определенных участках позволяет определить уровень развития пузырей на самом раннем этапе, что уже помогает с высокой долей вероятности определить тип буллезного эпидермолиза. Точку в диагностике этого состояния ставит генетический анализ методом прямого секвенирования генов, которые ассоциированы с тем или иным типом заболевания. Такой многостадийный подход к диагностике буллезного эпидермолиза обеспечивает высокую точность.
Значительно упростить диагностику этого заболевания позволяет изучение наследственного анамнеза пациента, по которому можно выявить его кровных родственников с такой же проблемой. Кроме того, если у кого-то из родных имеется буллезный эпидермолиз, имеет смысл производить пренатальную генетическую диагностику, что позволит выявить наличие данной патологии на ранних этапах развития плода. Дифференциальную диагностику осуществляют с истинной пузырчаткой, некоторыми формами буллезного пемфигоида, приобретенным буллезным эпидермолизом (который является не наследственным, а аутоиммунным заболеванием).
Лечение буллезного эпидермолиза
Специфического лечения этого заболевания не существует, все терапевтические процедуры сводятся к предупреждению развития осложнений и уменьшению выраженности пузырьков и эрозий. В случае тяжелых форм буллезного эпидермолиза назначают преднизолон. Из наружных терапевтических манипуляций производят асептическое вскрытие пузырьков, обработку их крышки антисептиками, накладывают гелиомициновую мазь. Наложение повязок нужно производить крайне осторожно, так как давление бинтов может спровоцировать появление новых пузырей. При наличии осложнений (шока, сепсиса) проводят симптоматическое лечение противошоковыми препаратами и антибиотиками. С профилактической целью можно производить облучение кожных покровов ультрафиолетовыми лучами.
Современная генетика и ряд других областей медицины продолжают широкие исследования буллезного эпидермолиза с целью поиска более эффективных методик лечения. Среди основных технологий и методов наиболее перспективными считаются способы с использованием стволовых клеток, белковая и генная терапии. Однако пока ни один из методов не вышел за рамки экспериментов на животных, поэтому буллезный эпидермолиз в настоящее время является неизлечимым заболеванием.
Прогноз буллезного эпидермолиза
Прогноз буллезного эпидермолиза чаще всего неопределенный, так как зависит от множества факторов и обстоятельств – типа заболевания, наличия или отсутствия у больного сопутствующих нарушений, его образа жизни. Например, локальный подтип простого эпидермолиза чаще всего имеет доброкачественное течение и редко создает угрозу жизни пациенту. Тогда как подтип Аллопо-Сименса имеет очень высокую смертность – как и от кожных проявлений, так и по причине отдаленных осложнений, таких как поражения почек и органов ЖКТ, а также развития плоскоклеточного рака кожи. Больные с такой проблемой должны бережно относиться к своей коже, не забывать про антисептическую обработку эрозий и других поражений, избегать занятий травмирующими видами спорта и иной деятельностью такого рода.
Фонд «Дети-Бабочки» совместно с Национальным медицинским исследовательским центром акушерства, гинекологии и перинатологии имени академика В.И. Кулакова запустили совместный проект по оказанию комплексной специализированной помощи детям с буллезным эпидермолизом и их семьям
На сегодняшний день буллезный эпидермолиз (БЭ) – неизлечимое заболевание с хроническим течением. Возможности современной медицины, по крайней мере пока, сводятся к симптоматической терапии, правильному уходу и поиску новых протоколов лечения, которые позволили бы лучше справляться с проявлениями БЭ, предотвращать (или хотя бы отодвинуть во времени) тяжелые осложнения и повысить качество жизни пациентов.
Уже почти 11 лет фонд «Дети-Бабочки» оказывает комплексную помощь пациентам с буллезным эпидермолизом и их семьям: обеспечивает медицинскую, психологическую и юридическую поддержку, медикаменты, патронаж, обучение врачей. Совместно с ФГБУ «Национальный Медицинский Исследовательский Центр акушерства, гинекологии и перинатологии имени академика В.И. Кулакова» Министерства здравоохранения Российской Федерации (ФГБУ «НМИЦ АГП им. В.И. Кулакова» МЗ РФ) фондом был инициирован специальный проект, который позволит детям с БЭ по всей России получить высококачественную медицинскую помощь.
Центр имеет большие диагностические мощности, активно использует инновационные технологии лечения, в частности, клеточную терапию. Фонд «Дети-Бабочки», в свою очередь, обладает организационными компетенциями и наработками в области ухода за детьми с буллезным эпидермолизом. Объединение усилий дает группе врачей и исследователей шанс на разработку новых протоколов лечения.
«Мы можем «подхватить» маленького пациента в самом начале его жизни и дать задел на будущее, ресурс для организма, чтобы как можно дольше избегать осложнений, таких как длительно незаживающие раны, срастание пальцев, контрактуры и тому подобное» – подчеркивает Юлия Юрьевна Коталевская, врач-генетик фонда.
В проекте участвуют дети с самой тяжелой дистрофической формой БЭ. Первый этап программы – проведение телемедицинского консилиума регионального врача и специалистов ФГБУ «НМИЦ АГП им. В.И. Кулакова» МЗ РФ. В процессе телеконсультации оценивается состояние новорожденного, согласовывается тактика медицинской помощи, определяется тактика ухода за кожей, обсуждается необходимость перевода ребенка в Москву. После дифференциальной диагностики ребенок госпитализируется в ФГБУ «НМИЦ АГП им. В.И. Кулакова» МЗ РФ, где разрабатывается индивидуальный план лечения.
Фонд финансирует собственно научную составляющую проекта, затраты на генетическую диагностику и медикаменты на весь период госпитализации. Пребывание в стационаре ребенка с мамой, необходимые исследования, лечение других заболеваний и уход производятся за счет средств ОМС. Транспортные расходы покрываются за счет региональных департаментов здравоохранения и средств благотворительного фонда «Дети-бабочки».
Программа пребывания в стационаре объединяет в себе две главные составляющие:
- правильный уход с первых дней жизни, который поможет изначально избежать серьезных (и, к сожалению, частых) ошибок в ведении пациентов с БЭ;
- терапию с применением клеточных технологий, направленную на стимуляцию процессов регенерации и поддержание стабильности кожного покрова.
Специалисты фонда курируют лечение на всех его стадиях и находятся в постоянном контакте с врачами в отделении и мамой пациента.
После поступления ребенку назначается комплексное обследование и консультация узкопрофильных специалистов. К моменту перевода пациента в отделение фонд организует доставку специальных перевязочных материалов. Дополнительно под контролем медиков маму обучают основам ухода за ребенком.
Изначально проект был рассчитан на три года, но пандемия «скорректировала» его продолжительность. Тем не менее, в 2022 году планируется публикация промежуточных данных, которые в первом приближении позволят сделать выводы об эффективности протоколов клеточной терапии.
Исследования по поиску эффективных способов борьбы с патологией кожи проводятся во всем мире. Совместный проект Фонда и ФГБУ «НМИЦ АГП им. В.И. Кулакова» МЗ РФ по созданию мультидисциплинарного подхода в сочетании с клеточными технологиями – один из шагов в этом направлении.

Буллезный эпидермолиз — редкое генетическое заболевание кожи, о котором зачастую не знают не только обычные люди, но и врачи. В начале Недели буллезного эпидермолиза предлагаем полный ликбез по этому заболеванию
Буллезный эпидермолиз — это пожизненная проблема, от которой нельзя избавиться по желанию, с помощью дорогостоящего лечения или иностранных врачей. Поэтому буллезный эпидермолиз оказывает на больного сильное психологическое воздействие: наличие видимых изменений кожи и частое посещение больниц могут повлиять на социальную, учебную и профессиональную жизнь человека, значительно снизив ее качество. Сказывается и не всегда корректная реакция незнакомых людей.
Кроме того, несмотря на развитость информационных технологий, далеко не все врачи в России сегодня знают о буллезном эпидермолизе и могут поставить правильный диагноз. В итоге многих «бабочек» (так называют пациентов с диагнозом) годами «лечат» от мнимой пищевой аллергии или других заболеваний, не имеющих к реальности никакого отношения.
Чем больше общественность, в том числе и профессиональная, будет знать о буллезном эпидермолизе и связанных с ним проблемах, тем больше внимания, принятия и квалифицированной медицинской помощи будут получать такие больные. В России Неделю буллезного эпидермолиза проводит Благотворительный фонд «Дети-бабочки».

Маргарита Гехт,
ведущий врач-дерматолог благотворительного фонда «Дети-бабочки»
Что такое буллезный эпидермолиз
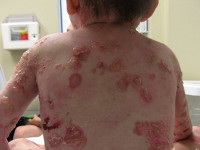
Буллезный эпидермолиз (БЭ) — не одно, а целая группа редких генетических заболеваний. У людей с таким диагнозом кожа и слизистые оболочки очень хрупкие. От малейшего прикосновения на них образуются пузыри. Лопаясь, они оставляют после себя болезненные раны.
Эти пузыри наверняка знакомы и вам. Такая влажная мозоль возникает на коже после целого дня, проведенного в тесных туфлях. Вечером вы нередко обнаруживаете ее уже вскрывшейся. Но если у обычных людей такие пузыри вздуваются из-за длительного трения тесной обуви и плотно прилегающей кожи, то у людей с БЭ — спонтанно или в результате малейших травм. Именно поэтому пациентов с этим заболеванием метафорично называют «бабочками». Как известно, даже легкое прикосновение к крылу бабочки снимает с него защитный слой, в результате чего она уже не может летать. При БЭ поврежденная кожа болит и может инфицироваться.
Чтобы понять, как возникает буллезный эпидермолиз, необходимо знать строение человеческой кожи.
Как устроена кожа
Кожа — самый большой орган нашего тела. Она защищает организм от микробов и аллергенов, помогает регулировать температуру тела и позволяет ощущать прикосновение, тепло и холод.
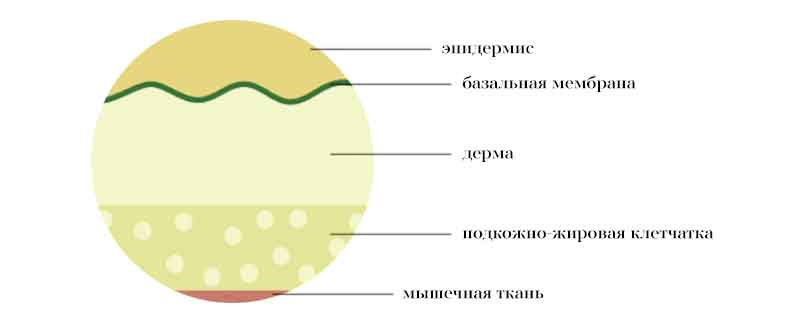
Кожа имеет три слоя:
Эпидермис
Внешний слой кожи, видимый глазу. Его основные функции — защитная и сохранения водонепроницаемого барьера — гидролипидной мантии.
Дерма
Этот слой находится под эпидермисом. Содержит плотную соединительную ткань, волосяные, сальные фолликулы, потовые железы, коллаген, эластин. В дерме проходят кровеносные сосуды и нервные окончания.
Между эпидермисом и дермой находится базальная мембрана — важная структура, соединяющая эти два слоя.
Гиподерма, или подкожно-жировая клетчатка
Глубокий слой кожи. Он состоит из жировой и соединительной ткани.
Работу, а главное, взаимодействие слоев кожи между собой определяют различные «помощники»: гены, ферменты, белки, дополнительные структуры и клетки. В случае нарушения работы хотя бы одного из «помощников» в коже происходят различные изменения. Так, при внезапно возникающих или вызванных искусственно стойких изменениях наследственных структур появляются генодерматозы, в том числе и буллезный эпидермолиз.
Что такое генодерматозы
Генодерматозы — наследственные заболевания кожи, насчитывающие несколько сотен нозологических форм и проявляющиеся различными патологическими процессами.
Причина их возникновения кроется в генах человека. Они хранят информацию, которая переводится в структуры кожи, содержащие белки, а именно — в кератин, коллаген, ламинин и интегрин, которые обеспечивают целостность и устойчивость кожи. При генетических поломках эти структуры перестают выполнять свою скрепляющую функцию, в результате кожа теряет целостность и становится хрупкой.
Буллезный эпидермолиз — один из самых тяжелых видов дерматозов данной группы.
Какие бывают виды буллезного эпидермолиза
Существует четыре основных формы буллезного эпидермолиза:
- простая;
- пограничная;
- дистрофическая;
- синдром Киндлера;
Классификация БЭ зависит от того, на каком уровне кожи произошел патологический процесс.
Простая форма
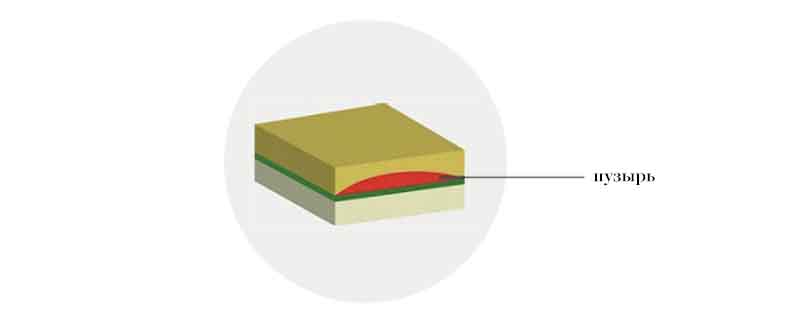
При данной форме БЭ пузыри появляются в пределах эпидермиса. Они не приводят к образованию рубцов, но причиняют сильную боль. Кроме того, летом, когда усиленное потоотделение провоцирует образование новых пузырей, простая форма БЭ всегда протекает тяжелее.
Пограничная форма
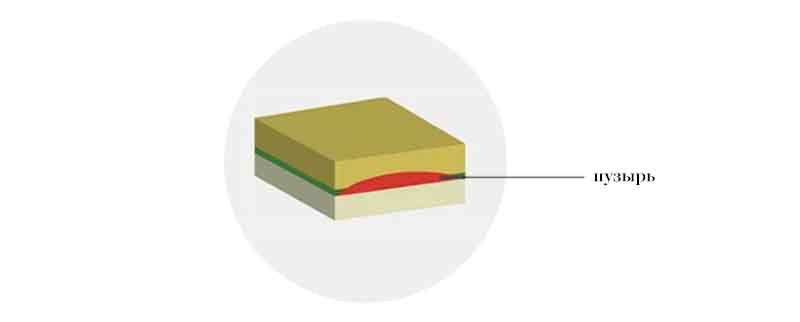
В данном случае расщепление происходит внутри базальной мембраны — структуры, которая «скрепляет» эпидермис и дерму. Более того, при определенном подтипе такой формы базальная мембрана вообще отсутствует. Из-за этого одна кожная структура может «находить» на другую, в результате чего на коже возникают многочисленные пузыри и обширные раны. Сливаясь, раны часто не оставляют на коже живого места и могут вызвать сепсис, а значит, потенциально опасны для жизни.
Дистрофическая форма
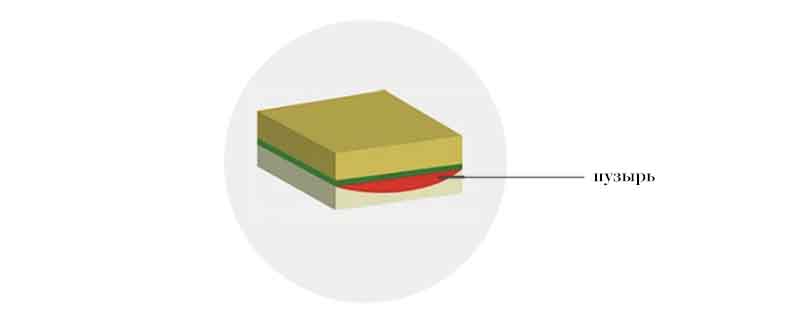
При данном типе БЭ расщепление происходит ниже базальной мембраны, в дерме. В этой области залегают ключевые структуры, которые определяют плотность и упругость кожи, — коллаген и эластин. При такой форме БЭ патологический процесс протекает именно в коллагене, в результате чего пузыри заживают с последующим образованием рубцовой ткани.
Синдром Киндлера
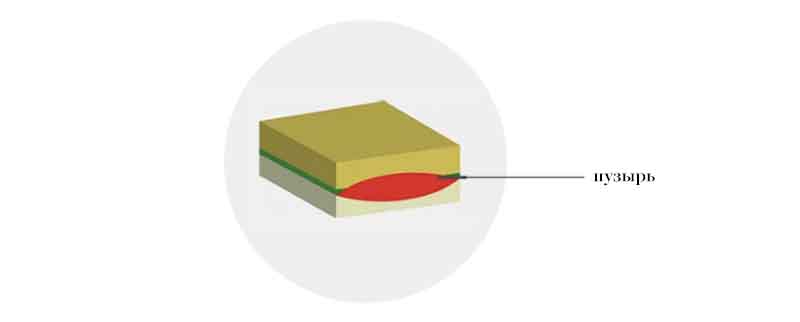
В этом случае пузыри могут образовываться одновременно на разных уровнях кожи. Кожные проявления могут быть вариабельны и зависят от степени вовлеченности гена в мутацию.
Чем отличаются врожденный и приобретенный БЭ
БЭ можно разделить на две группы — врожденный и приобретенный.
Врожденный БЭ, которому посвящена большая часть данной статьи, обусловлен генетическим дефектом. Этот дефект возникает еще внутриутробно, а сразу после рождения становятся видны внешние клинические проявления — раны на коже. Сегодня эта форма БЭ не имеет патогенетического лечения.
Приобретенный БЭ — это аутоиммунный дерматоз, связанный с выработкой специфических антител к собственному коллагену VII типа — наиболее важному компоненту фибрилл. Фибриллы — особые структуры в форме якоря, которые вместе с другими компонентами кожи скрепляют ее слои между собой.
При приобретенном БЭ образование пузырей происходит после связывания аутоантител — это антитела, которые выработались на собственные белки в результате воздействия на организм триггерных факторов (среди которых можно выделить стресс, перенесенные заболевания и оперативные вмешательства с коллагеном VII типа). Это, в свою очередь, приводит к активации каскада воспалительных реакций со стороны кожи.
Клинически заболевание характеризуется натянутыми пузырями на травмированных участках тела, которые заживают с образованием рубца. Так проявляется механобуллезная форма приобретенного БЭ. Второй наиболее частый подтип приобретенного БЭ — буллезное пемфигоидоподобное заболевание. Для него характерен кожный зуд и поражение слизистых оболочек. Для лечения приобретенного БЭ назначают местное и системное использование глюкокортикоидов и введение внутривенных иммуноглобулинов.
Как диагностируется буллезный эпидермолиз
Детальная лабораторная диагностика необходима для определения формы и подтипа БЭ и точной причины его возникновения на генетическом и белковом уровнях. Это помогает прогнозировать тяжесть состояния, оказывать надлежащую медицинскую помощь и участвовать в клинических испытаниях.
В лабораторной диагностике БЭ используются два основных метода:
1. Генетическое тестирование, которое направлено на выявление в ДНК мутации в конкретном гене, вызывающей заболевание.
2. Анализ образцов кожи с использованием методов, работающих на уровне белка, который позволяет выявить изменения в белке и компонентах кожи.
Влияет ли буллезный эпидермолиз только на кожу
Хотя физические эффекты БЭ затрагивают в основном кожу, при некоторых формах БЭ поражается не только кожа, но и слизистые оболочки внешних и внутренних органов: глаз, полости рта, пищевода, кишечника.
Заразен ли буллезный эпидермолиз
БЭ — генетическое заболевание. Это значит, что с ним рождаются. Заразиться буллезным эпидермолизом нельзя.
Как наследуется это заболевание
БЭ может быть унаследован одним из трех способов:
- аутосомно-доминантным наследованием;
- аутосомно-рецессивным наследованием;
- наследованием de novo.
Аутосомно-доминантное наследование
В этом случае один из родителей страдает заболеванием и имеет дефектный ген. Он передает измененный ген своему ребенку. Вероятность того, что любой их ребенок родится с БЭ, составляет 50%.
Аутосомно-рецессивное наследование
В этой ситуации оба родителя клинически здоровы и не имеют внешних проявлений заболевания, но являются его генетическими носителями. Чтобы их ребенок родился с БЭ, он должен унаследовать дефектный ген — часть гена от матери, часть — от отца. Возникает всем известная математическая комбинация, когда минус на минус дает плюс в виде болезни у ребенка. Вероятность того, что это произойдет, составляет 25%.
Мутации, вызывающие БЭ, которые происходят de novo
Человек, несущий вариант de novo, то есть родившийся с БЭ при здоровых родителях, в том числе не имеющих генных мутаций, имеет 50%-й шанс передать заболевание своим детям.

Можно ли вылечить БЭ
На сегодняшний день нет эффективного лечения ни одной из форм БЭ. Все доступное лечение направлено на облегчение симптомов, чтобы, в конечном счете, улучшить качество жизни человека с данным генодерматозом.
Какова продолжительность жизни пациентов с БЭ
Продолжительность жизни при буллезном эпидермолизе зависит от формы заболевания, качества ухода за кожей, профилактики внекожных проявлений, качества оказываемой медицинской помощи и прохождения ежегодных обследований. При соблюдении этих условий пациенты могут жить полноценной и качественной жизнью. В таких случаях ее продолжительность близка к продолжительности жизни людей без особенностей здоровья.
Что включает в себя облегчение симптомов БЭ
Симптоматическое лечение имеет важнейшее значение для людей с БЭ. Обычно оно включает ежедневный уход за непораженной кожей, за кожей с пузырями и ранами, а также обезболивание.
Пациентам с более тяжелыми типами БЭ требуются дополнительные процедуры, в том числе прием препаратов железа, переливание эритроцитарной массы и альбумина — белка, находящегося у данных пациентов в большом дефиците. Именно от его количества в организме зависит скорость заживления кожи, в том числе и после хирургических вмешательств. Пузыри поражают слизистую и заживают только через образование рубца, который сужает просвет органа или ограничивает его функцию. Хирургические операции позволяют освободить сросшиеся пальцы и открыть пищевод для нормального поступления пищи.
Как выглядит симптоматическое лечение при БЭ
Характер и стоимость ухода за ранами зависят от многих факторов, в том числе имеют значение:
- форма БЭ;
- возраст пациента;
- общее восприятие;
- текущее состоянии кожи;
- состояние питания;
- домашнее окружение;
- наличие качественных перевязочных материалов.
Основное лечение подразумевает использование атравматического материала, с которым при снятии не будет отходить кожа.
Основные этапы перевязки:
Тщательная подготовка к перевязке со всеми необходимыми материалами и доступным механизмом утилизации старого перевязочного материала сокращает длительность перевязки и снижает риск возникновения инфекции.
Все лица, участвующие в смене повязок, должны в обязательном порядке проводить предварительную дезинфекцию рук.
Оценка состояния кожи
Все пузыри на коже необходимо вскрывать, затем очищать образовавшиеся раны, при инфицировании — лечить.
При перевязке на кожу сначала накладывают сетчатую или губчатую повязку, затем для впитывая экссудата и излишков крема — промежуточную, после этого — фиксирующую повязку.

Что важно при выборе перевязочных средств и уходе за кожей при БЭ
Интенсивность ухода за ранами определяется в первую очередь формой БЭ. При простой форме, когда пузыри и раны возникают на отдельных участках тела, а не повсеместно, перевязка проходит гораздо быстрее, чем при более тяжелой дистрофической форме, требующей обработки повязками большей части кожи.
К счастью, в настоящее время есть множество перевязочных средств и медикаментов, которые подходят для ухода за ранами при буллезном эпидермолизе. Однако найти «правильный» материал все еще нелегко. То, что подходит для одного больного, может оказаться совсем неподходящим для другого. Поэтому даже при одной и той же форме БЭ перевязочный материал и уходовые средства подбираются индивидуально. Оценка такого соответствия должна всегда производиться совместно с врачом.
При выборе перевязочных средств необходимо также учитывать их доступность на местном уровне, ведь в разных странах для этих целей разработаны и доступны разные продукты. Кроме того, на перевязочные средства требуется значительная сумма. Так, в России на уход за кожей человека-бабочки ежемесячно нужно от 50 до 400 тыс. руб. Сегодня наше государство не всегда покрывает эти траты, а при обеспечении за счет бюджета отдает предпочтение менее дорогостоящим материалам.
Еще одним важным аспектом в лечении больного БЭ является гигиена и уход за участками кожи с рубцами, без пузырей и ран. Такой коже в первую очередь требуются восстановление и увлажнение. Чем лучше и качественнее увлажнена кожа, тем меньше выражены рубцы, меньше зуда и ниже риск инфицирования. Увлажняющие крема необходимо наносить на открытые участки кожи два-три раза в день.
Может ли легкая форма буллезного эпидермолиза перейти в тяжелую
Нет, поскольку причины, вызывающие одну форму БЭ, отличаются от причин возникновения других типов этого заболевания. Они, по сути, являются отдельными состояниями, поэтому одна форма не может перерасти в другую.
Читайте также: