
Витилиго наследуется как аутосомно доминантный признак
Обновлено: 19.04.2024
Для цитирования: Дворянкова Е.В., Корсунская И.М. Современная концепция наследуемости витилиго // Эффективная фармакотерапия. 2020. Т. 16. № 9. С. 32–35.
- Аннотация
- Статья
- Ссылки
- Английский вариант
Витилиго – аутоиммунный гипомеланоз со сложным механизмом наследования. На сегодняшний день определено 54 локуса генов, ответственных за его развитие. Согласно результатам современных исследований, наследуемость отмечается у 40% пациентов. Однако генетический фактор присутствует приблизительно в 80% случаев. Остальные 20% случаев предположительно обусловлены воздействием внешних факторов.
Дальнейшее исследование модели генетической обусловленности витилиго послужит инструментом для изучения других аутоиммунных заболеваний с полигенным риском наследования и развития персонализированной медицины.
- КЛЮЧЕВЫЕ СЛОВА: витилиго, генетическая обусловленность, аутоиммунный синдром, дерматоз
Витилиго – аутоиммунный гипомеланоз со сложным механизмом наследования. На сегодняшний день определено 54 локуса генов, ответственных за его развитие. Согласно результатам современных исследований, наследуемость отмечается у 40% пациентов. Однако генетический фактор присутствует приблизительно в 80% случаев. Остальные 20% случаев предположительно обусловлены воздействием внешних факторов.
Дальнейшее исследование модели генетической обусловленности витилиго послужит инструментом для изучения других аутоиммунных заболеваний с полигенным риском наследования и развития персонализированной медицины.
Витилиго – одно из первых заболеваний кожи, описанное в «Трактате о цвете кожи человека» французским хирургом Клодом-Николя Ле Кат (1765 г.). Несмотря на столь давнее знакомство медицинского сообщества с данным дерматозом, причины и механизмы его развития остаются недостаточно изученными.
Генетическое подтверждение аутоиммунной теории развития
Одной из основных теорий развития витилиго является генетическая обусловленность. Впервые о наследовании витилиго было сообщено в 1950 г.: G. Stüttgen и H. Teindel одновременно описали случаи развития заболевания у нескольких близких родственников в нескольких семьях [1, 2]. В частности, G. Stüttgen отметил, что помимо витилиго несколько членов наблюдаемых семей страдали аутоиммунными заболеваниями щитовидной железы. Он также предположил, что наследование витилиго может зависеть как от доминантных, так и от рецессивных факторов.
В середине 1960-х гг. появились новые методы изучения генетической основы витилиго и выявления генов, обусловливающих его развитие.
На сегодняшний день с помощью метода полигеномного поиска ассоциаций (Genome-Wide Association studies, GWA study – GWAS), позволяющего установить связь между геномными вариантами и фенотипическими признаками, определено 54 локуса генов, ответственных за развитие витилиго. В данном случае речь идет о методе поиска связи между однонуклеотидными полиморфизмами и заболеваниями человека.
В ходе таких исследований помимо генетических факторов риска развития витилиго были определены биологические основы восприимчивости к болезни. Это в свою очередь позволило начать разработку новых методов лечения.
Установлено, что большинство выделенных локусов генов являются регулярными [4]. Так, варианты с наибольшим общим влиянием на риск развития витилиго в масштабах всего генома находятся на хромосоме 6, в областях основного комплекса гистосовместимости I и II классов [5, 6]. Около 85% идентифицированных генов кодируют белки, участвующие в иммунных реакциях и апоптозе [7].
Кроме того, в ходе независимых генетических исследований других аутоиммунных заболеваний было идентифицировано около половины генов, которые эпидемиологически связаны с витилиго, что также поддерживает концепцию общей аутоиммунной предрасположенности к развитию данного гипомеланоза [8].
Наследуемость
Необходимо отметить, что при применении разных аналитических подходов для оценки риска развития витилиго вследствие генетической изменчивости (наследуемости) учитывалось, что другим фактором может быть воздействие окружающей среды. Большинство аналитических подходов основаны на оценке наличия заболевания у членов семьи больного либо в парах близнецов. Эти данные сравнивали с генетическими маркерами, полученными от субъектов без витилиго [9]. Количество таких субъектов варьировалось от сотен тысяч до нескольких миллионов. Такой анализ позволяет получить реальные величины наследуемости конкретного заболевания. Установлено, что в европейской популяции общая наследуемость витилиго достигает практически 40% [10], среди близнецов – 75–84% [7], в зависимости от типа изучаемых родов. В других популяциях встречаемость данного дерматоза среди близнецов колебалась от 50 до 80% [10, 11].
Полученные результаты свидетельствуют, что различие по заболеваемости в разных популяциях невелико. Однако она выше среди близнецов. Аналогичный показатель для других заболеваний с полигенным наследованием в данной когорте составляет 30–50% [12].
Таким образом, в европейской популяции риск развития витилиго в 80% случаев связан с генетическими факторами. Остальные 20% случаев наследуемости, а также 23% общего риска развития витилиго приходятся на редкие варианты (частота минорных аллелей от 0,01 до 0,0001).
Согласно результатам последних исследований, общие генетические варианты (частота обнаружения аллелей риска – более 0,01) составляют около 71% общей наследуемости витилиго и около 53% общего риска развития витилиго, причем значительная доля приходится на 50 общегеномных значимых локусов, идентифицированных с помощью GWAS [6].
Полученные данные позволяют предположить, что генетическая архитектура витилиго может быть менее полигенной, чем у других генетически сложных заболеваний.
Оценка риска полигенного наследования витилиго имеет высокую прогностическую ценность. Относительный риск (ОР) составил 8,79, что существенно выше, чем при оценке полигенного риска других генетически сложных заболеваний, таких как ишемическая болезнь сердца (ОР – 4,83), сахарный диабет 2-го типа (ОР – 3,30), воспалительные заболевания кишечника (ОР – 3,87), рак молочной железы (ОР – 3,36) [13].
Крупномасштабные эпидемиологические исследования наследуемости показали, что подавляющее большинство (91%) клинических случаев являются симплексными, то есть когда близкие родственники, страдающие витилиго, неизвестны, около 9% – мультиплексными семейными кластерами, когда известно несколько близких родственников с данной патологией [14]. Полигенный риск витилиго у пробандов таких мультиплексных семейств значительно выше, чем в симплексных случаях. Кроме того, в мультиплексных семьях общий полигенный риск развития заболевания наиболее часто передается от родственников с визуально неизмененной кожей, без явных признаков депигментирующего заболевания.
Таким образом, полигенное наследование витилиго выявляется как в симплексных, так и в мультиплексных случаях, с участием одних и тех же локусов генов, которые были обнаружены с помощью GWAS преимущественно в симплексных случаях. Подавляющее большинство мультиплексных семейств несут и передают большое количество общих аллелей предрасположенности к заболеванию, но с относительно низким процентом случаев его клинического проявления.
Особенности дебюта
Витилиго считается заболеванием молодых, так как его дебют приходится преимущественно на возраст моложе 20 лет. Однако эпидемиологический анализ, проведенный в европейской популяции, свидетельствует о бимодальном распределении дебюта витилиго [6], так же как при некоторых других аутоиммунных заболеваниях, например при сахарном диабете 1-го типа и ревматоидном артрите.
Согласно результатам эпидемиологического исследования возраста начала витилиго у европейцев, в последнее время наблюдается значимая тенденция к увеличению такового [15]. Ретроспективно были проанализированы данные 4406 больных с 1951 по 2013 г. Средний возраст дебюта за указанный период повысился в два раза. Изменение было наиболее выраженным с 1973 и 2000 г. Так, до 1970 г. начало заболевания приходилось на возраст 14,6 ± 9,4 года. С 1973 г. возраст дебюта витилиго увеличивался примерно на четыре месяца в год и стабилизировался только в 2004 г. Таковой составлял 30,2 ± 17,3 года. Аналогичная ситуация прослеживалась как в Европе, так и в Северной Америке. Поскольку генетика не изменилась, возрастной сдвиг дебюта заболевания свидетельствовал о том, что с 1973 и 2000 г. либо уменьшилось воздействие триггерных факторов, либо изменилась биологическая реакция на них. Наиболее очевидной причиной, по мнению авторов исследования, является увеличивающаяся из года в год частота применения солнцезащитных средств.
Последние данные указывают на то, что средний возраст дебюта витилиго составляет 25,9 ± 16,6 года и, по-видимому, включает две пересекающиеся возрастные группы: ранняя группа (около 38% случаев) – средний возраст начала заболевания 10,3 ± 5,6 года, поздняя группа (примерно 62% случаев) – 34,0 ± 14,5 года [6]. Отдельные генетические анализы в данных группах показали, что известные локусы, определенные с помощью GWAS, вносят одинаковый вклад в развитие заболевания. Тем не менее стратифицированный анализ, проведенный в ранней группе, позволил выявить новую, очень сильную специфическую ассоциацию с необычным инсерционно-делеционным полиморфизмом в области II класса MHC на хромосоме 6, так называемый гаплотип МНС enhancer. Гаплотип, который содержит два варианта MHC enhancer, специфически связан с очень высоким риском развития витилиго (ОР – 8,1) и ранним началом заболевания, преимущественно в возрасте от пяти до девяти лет.
Функциональные исследования доказали, что гаплотип раннего витилиго высокого риска ассоциируется с повышенной экспрессией как мРНК HLA-DQB1, так и общего белка HLA-DQ на моноцитах и дендритных клетках в периферической крови. Повышенная экспрессия HLA II класса может способствовать презентации триггерных антигенов на поверхности этих антигенпрезентирующих клеток, увеличивая вероятность аутореактивной активации Т-клеток.
Расширенный гаплотип MHC enhancer с высоким риском раннего развития также несет классические аллели HLA: HLA-DRB1*13:01, HLA-DRB3*01:01, HLA-DQA1*01:03, HLA-DQB1*06:03. Генетический анализ свидетельствует, что сами по себе данные классические аллели HLA не обусловливают риск развития витилиго. Скорее всего, они являются некодирующей вариацией enhancer, которая и предрасполагает к возникновению витилиго. Это согласуется с повышенной экспрессией гена HLA-DQB1 при раннем дебюте заболевания. Тем не менее распространенность других ассоциированных аутоиммунных заболеваний в ранней группе была ниже, чем в поздней группе, – 17,0 против 22,4% случаев.
При этом HLA-DRB1*13:01, который находится на гаплотипе MHC II класса, ассоциированном с очень высоким риском развития раннего витилиго, был связан с очень низким риском развития системной красной волчанки, ревматоидного артрита и сахарного диабета 1-го типа. Таким образом, рассматриваемый гаплотип может одновременно создавать высочайший риск для развития витилиго с ранним началом и обеспечивать относительную защиту от возникновения других аутоиммунных заболеваний, которые часто эпидемиологически связаны с данным гипомеланозом.
Факторы риска
Как было указано выше, в 80% случаев развитие витилиго обусловлено генетически. В 20% случаев оно является следствием воздействия негенетических факторов, среди которых наиболее распространенным и доказанным является феномен Кебнера. Предполагают, что повреждение кожи, возможно осложненное субклинической инфекцией, играет ключевую роль в инициации витилиго. Однако до сих пор неизвестно, индукция витилиго способна включать только один триггер или несколько. В последнем случае кумулятивный эффект с течением времени может привести к потере толерантности к аутоантигенам меланоцитов.
За последние пять лет достигнут значительный прогресс в понимании биологических основ витилиго. Это позволило с достаточной точностью оценить относительное влияние генов и факторов окружающей среды на риск развития заболевания. Генетический риск развития заболевания как на фоне общих, так и на фоне редких вариантов наследования, по-видимому, является аддитивным и полигенным по своей природе, и риск от 50 общих вариантов, идентифицированных с помощью GWAS, может быть объединен в группу полигенного риска, которая в некоторой степени уже может предсказать возможность развития витилиго у конкретного индивида. Прогностическая эффективность оценки полигенного риска витилиго, несомненно, требует дальнейшей разработки и может существенно повыситься с открытием и включением в анализ дополнительных вариантов риска.
До настоящего времени не были точно определены генетически обусловленные подгруппы витилиго, которые имеют разную базовую полигенную патобиологию. Возможность правильно распределять конкретные клинические случаи витилиго в генетически определенные полигенные подгруппы способна улучшить определение специфичных для подгрупп показателей полигенного риска, которые включают локусы, соответствующие каждой подгруппе.
Все локусы генов, ответственные за развитие витилиго, были выявлены путем сравнения биологического материала, полученного от больных европейцев, с контрольными образцами, взятыми у лиц с визуально неизмененной кожей либо у ближайших родственников больных без признаков данной патологии. Таким образом, все эти локусы ассоциированы со статусом «случай против контроля» или, для гаплотипа MHC высокого риска, с ранним началом витилиго. Однако до сих пор неизвестно, влияет ли какой-либо из этих локусов или все они на клиническое течение витилиго и прогноз заболевания.
Важно отметить, что оценка полигенного риска развития витилиго, основанная на результатах генетических исследований суперпопуляции европеоидной расы, может быть неприменима к другим суперпопуляциям, в которых могут преобладать иные этиологически значимые локусы и аллели.
В настоящее время витилиго расценивается как заболевание с полигенным наследованием множественных аллелей риска, которые, однако, вызывают относительно небольшие клинические проявления. Поэтому рассматривать только вариант полигенного наследования не представляется верным. Редкие случаи развития витилиго могут быть результатом передачи только одного или нескольких редких локусов, которые еще не были обнаружены, как это имеет место при редких менделевских множественных аутоиммунных синдромах, к которым иногда относят витилиго. Соответственно геномный анализ мультиплексных семейств больных витилиго, которые передают низкий полигенный риск развития заболевания посредством известных локусов, может позволить обнаружить редкие либо частные варианты, которые имеют чрезвычайно высокий генетический риск. И наоборот, некоторые мультиплексные семьи с низким полигенным риском могут подвергаться воздействию необычного либо чрезвычайно сильного внешнего фактора. В любом случае исследование причин возникновения семейных кластеров с явно низким полигенным риском развития витилиго представляется многообещающим для установления новых генетических и негенетических причин развития заболевания.
На сегодняшний день получено недостаточно данных о факторах экологического риска или триггерах витилиго. Прежде всего это связано с трудностями в реализации надежных методов их выявления, регистрации и отслеживания. В то же время тенденция к повышению возраста дебюта витилиго при неизменности генетических триггеров свидетельствует о значимом влиянии потенциальных экологических триггеров.
Учитывая, что наследуемость витилиго довольно высока, риск витилиго в основном полигенный (54 локуса, выявленные с помощью GWAS), оценка полигенного риска, аддитивно сочетающая эти локусы, достаточно точна, данное заболевание можно считать идеальной моделью для проверки общих гипотез генетической архитектуры и наследования сложных заболеваний.
Оценка полигенного риска развития витилиго была использована для изучения природы наследования сложных заболеваний в мультиплексных семьях по сравнению с симплексными случаями. В обоих отмечен полигенный риск из одних и тех же локусов.
Таким образом, витилиго можно рассматривать как модель для исследования генетической архитектуры ряда болезней, а также для прогностических аспектов персонализированной медицины.
Задача 1: Подагра определяется доминантным аутосомным аллелем гена. Пенетрантность гена у мужчин составляет 20%, а у женщин равна 0 %. Какова вероятность заболевания подагрой в семье гетерозиготных родителей?
Решение:
Р: ♀ Аа - ♂ Аа
G: А а А а
F1 АА : 2Аа : аа
Вероятность того, что в семье появятся дети, несущие аллель гена подагры, равна 75%. Но признак проявится лишь у мужчин. Вероятность рождения мальчиков – 50%. Следовательно, ген подагры наследуется в
75% × 50% / 100% = 37,5%.
Ген подагры проявится лишь у 20% несущих его мужчин:
37,5% × 20% / 100% = 7,5%
Ответ: Вероятность заболевания подагрой в этой семье составит 7,5%.
Задача 2: По данным шведских генетиков одна из форм шизофрении наследуется как доминантный аутосомный признак. У гомозигот пенетрантность – 100%, у гетерозигот – 20%. Определите вероятность заболевания детей в семье, где один из супругов гетерозиготен, а другой нормален в отношении анализируемого признака? Определите вероятность рождения больных детей в браке двух гетерозиготных родителей.
Решение:
По условию задачи некоторые формы шизофрении наследуются как доминантный аутосомный признак с неполной пенетрантностью. В первом случае один из супругов нормален в отношении анализируемого признака, а другой гетерозиготен. Тогда, обозначив ген, определяющий шизофрению, А, мР: ♀ Аа - ♂ аа
G: А а а
F1 Аа : аа
1/2 больные: 1/2 здоровые
Отсюда вероятность рождения ребенка, несущего ген шизофрении, равна 1/2. У гетерозигот пенетрантность признака составляет 20% или 1/5. Перемножив вероятность носительства гена на вероятность его проявления, получим: 0,5×0,2=0,1 или 10%.
Во втором случае имеет место брак двух гетерозиготных индивидов. В таком браке:
Р: ♀ Аа - ♂ Аа
G: А а А а
F1 АА : 2Аа : аа
Вероятность рождения гомозиготы АА — 1/4, вероятность рождения гетерозиготного ребенка — 1/2. Пенетрантность гена у гомозигот равна 100%, то есть все они будут больны шизофренией. Для гетерозигот пенетрантность — 20% или 1/5. Больные дети могут появиться с вероятностью: 0,5×0,2=0,1. В итоге вероятность рождения больного ребенка в таком браке будет: 0,25 + 0,1 =0,35 или 35%.
Ответ: В первом случае вероятность рождения больного ребенка 10%, а во втором — 35%.
ЗАДАЧИ:
1. Отосклероз наследуется как доминантный аутосомный признак с пенетрантностью 30%. Отсутствие боковых верхних резцов наследуется как сцепленный с Х-хромосомой рецессивный признак с полной пенетрантностью. Определите вероятность проявления у детей обеих аномалий одновременно в семье, где мать гетерозиготна в отношении обоих признаков, а отец нормален по обеим парам аллелей генов.
2. Карий цвет глаз доминирует над голубым и определяется аутосомным аллелем гена. Ретинобластома определяется другим доминантным аутосомным аллелем гена. Пенетрантность ретинобластомы составляет 60%. Какова вероятность того, что здоровыми от брака гетерозиготных по обоим признакам родителей будут кареглазые дети?
3. Арахнодактилия наследуется как доминантный аутосомный признак с пенетрантностью 30%. Леворукость– рецессивный признак с полной пенетрантностью. Определить вероятность проявления обеих аномалий одновременно у детей в семье, где оба родителя гетерозиготны по обеим парам генов.
4. Черепно-лицевой дизостоз (преждевременное зарастание швов черепа и незаращение большого родничка) наследуется как аутосомно-доминантный признак с пенетрантностью 50%. Определите вероятность рождения больного ребенка, если один из родителей гетерозиготен по данному заболеванию, а другой здоров.
5. Синдром Ван-дер-Хеве (голубая окраска склер, ломкость костей, глухота) имеет аутосомно-доминантный тип наследования. Пенетрантность этих признаков изменчива. По данным К. Штерна (1965) она составляет для голубых склер 100%, по ломкости костей – 63%, по глухоте – 60 %. Мужчина, имеющий голубой цвет склер и нормальный в отношении двух других признаков, вступил в брак со здоровой женщиной, в родословной которой случаев этого синдром не встречалось. Определите вероятность рождения в этой семье детей с признаками ломкости костей, если известно, что отец мужа данный синдром имел.
6. Ретинобластома (опухоль сетчатки глаза) обусловлена доминантным аллелем гена, пенетрантность которого составляет 70%. В медико-генетическую консультацию обратилась беременная женщина. Из анамнеза известно, что она и ее супруг здоровы, но имеют больного старшего сына. В семье женщины случаев ретинобластомы не было, а отец супруга в детстве был оперирован по поводу ретинобластомы. Какова вероятность рождения больного ребенка в этой семье?
7. Ангиоматоз наследуется как доминантный аутосомный признак с пенетрантностью 50%. Определите вероятность заболевания детей в семье, где оба родителя являются гетерозиготными носителями ангиоматоза.
Задача. Витилиго (нарушение пигментации) наследуется как аутосомно-доминантный признак с пенетрантностью 70 %. Какова вероятность рождения ребенка с витилиго, если у одного из родителей имеется витилиго?
Обозначим ген витилиго «А», а нормальный ген – «а», тогда страдать витилиго могут организмы с генотипами АА и Аа. Поэтому нужно рассмотреть 2 варианта скрещиваний.
1 вариант:
Вероятность появления генотипа Аа в первом поколении составляет 100 %. Имея в виду, что пенетрантность доминантного гена 70 %, можно сделать вывод, что вероятность рождения в семье ребенка с витилиго составит 70 %.
2 вариант:
 |  |
Все потомки с генотипом аа (50 %) оказываются здоровыми. Потомки с генотипом Аа также рождаются с вероятностью 50 %, но только 70 % из них будут с витилиго. Следовательно, вероятность рождения ребенка с витилиго – 35 %.
Задача 1. Подагра определяется доминантным аутосомным геном. По некоторым данным (Эфроимсон В.П., 1968) пенетрантность гена у мужчин составляет 20%, а у женщин она равна нулю. а) Какова вероятность заболевания подагрой в семье гетерозиготных родителей? б) Какова вероятность заболевания подагрой в семье, где один из родителей гетерозиготен, а другой нормальный по анализируемому признаку?
Задача 2. Ангиоматоз сетчатки глаза обусловливается доминантным аутосомным геном, пенетрантность которого 50 %. Какова вероятность (в процентах) рождения больного ребенка в семье, где оба супруга гетерозиготны по данному гену?
Задача 3. Отосклероз (очаговое заболевание косточек среднего уха, способное вызвать глухоту) наследуется как доминантный аутосомный признак с пенетрантностью 30%. Гипертрихоз (вырастание волос на краю ушной раковины) наследуется как признак, сцепленный с Y-хромосомой с полным проявлением к 17 годам. Жена здоровая и гомозиготна по исследуемому признаку, муж имеет обе аномалии. Какова вероятность рождения детей с отосклерозом?
Задача 4. Отосклероз (очаговое заболевание косточек среднего уха, способное вызвать глухоту) наследуется как доминантный аутосомный признак с пенетрантностью 30%. Отсутствие боковых резцов наследуется как сцепленный с Х-хромосомой рецессивный признак с полной пенетрантностью. Определите вероятность рождения детей с обеими аномалиями одновременно в семье, где мать гетерозиготна в отношении обоих признаков, а отец не страдает отсутствием резцов, но гетерозиготен по гену отосклероза.

Новость
Сильнее всего витилиго заметно, конечно, на тёмной коже, потому что контраст между пигментированными и «обесцвеченными» участками будет максимальным
Автор
Редакторы
Существуют не только заболевания, всерьёз угрожающие жизни, но и болезни, наносящие урон скорее имиджу человека, нежели его здоровью. К числу таких недугов относится витилиго — «загадочная болезнь», при которой поверхность кожи покрывают белые (лишённые пигментации) пятна, со временем увеличивающиеся и сливающиеся между собой. О природе этого явления известно очень мало, — в основном, только то, что оно имеет отношение к аутоиммунным процессам. Международный консорциум учёных провёл крупномасштабное генетическое сканирование, которое выявило несколько генов, с неправильной работой которых может быть связано развитие болезни. Правда, это пока лишь первый шаг — о лечении и даже о точных молекулярных и клеточных механизмах исследователи пока сказать ничего не могут.
Витилиго (от лат. vitiligo — накожная болезнь, лишай) — хроническое заболевание, выражающееся в первую очередь в появлении участков депигментированной кожи, волосяной покров в которых также становится седым. Это явление вызвано нарушением работы меланоцитов — клеток, производящих пигменты кожи (в первую очередь, меланин), — из-за их дисфункции или даже просто гибели. Витилиго имеет аутоиммунную природу — то есть, меланоциты гибнут из-за сбоя распознавания в системе клеточного иммунитета, когда свои собственные «тканевые стражи» (макрофаги, лимфоциты) начинают атаковать меланоциты и выводить их из строя. Витилиго часто сопровождается другими аутоиммунными заболеваниями, — например, тиреоидитом или системной красной волчанкой. Предварительные исследования показали, что генетические дефекты, вызывающие витилиго, в основном относятся к генам иммунной системы, а также к генам самих меланоцитов.

Рисунок 1. Знаменитая белая перчатка с блёстками поп-короля Майкла Джексона — видимо, лишь попытка замаскировать прогрессирующее развитие витилиго
Это заболевание, в общем-то, не опасно, но оно портит внешность человека. Широко известна история, когда поп-король Майкл Джексон, первоначально темнокожий, постепенно превратился в «белого человека» (рис. 1). Эта метаморфоза долго обсасывалась бульварной прессой, но, по-видимому, превращение было вызвано не расовыми убеждениями или чем-то более экстравагантным, а именно витилиго, первые признаки которого появились у певца в начале 1980-х. Знаменитая белая перчатка с блёстками, ставшая объектом подражания многочисленных имитаторов, служила для сокрытия начинающейся болезни, когда ладонь темнокожего артиста стала белеть. Позже, когда перчатка и грим перестали помогать, Джексон делал пластические операции для выравнивания общего оттенка кожи, — на тот момент, уже почти белой. В диагнозе «короля» значится также и системная красная волчанка.
Лечения витилиго не существует; есть лишь частичные меры, позволяющие замедлить развитие заболевания или уменьшить его внешние проявления. Прежде всего, больным необходимо использовать сильные солнцезащитные кремá, поскольку кожа, лишённая естественного фотофильтра, очень быстро сгорает на солнце, и в ней под действием ультрафиолетовых лучей даже может начать развиваться онкология. Впрочем, ультрафиолет диапазонов A и B используют и для терапии витилиго, но, конечно, в контролируемых клинических условиях. Кроме обычных косметических составов, лишь выравнивающих тон кожи, часто используют мази с кортикостероидами, которые в ряде случаев могут частично восстановить пигментацию. Кроме того, известны случаи удачной терапии путём «подсаживания» в поражённую область «своих» меланоцитов с участка здоровой кожи, размноженных в искусственных условиях. Помимо этого, есть данные об излечении болезни массой препаратов, полученных из природных источников — чёрного перца, гинкго и даже человеческой плаценты, но эти результаты нельзя назвать общепринятыми и широко распространёнными в медицинской и косметической практике.
Очевидно, что по-настоящему эффективного способа лечения не будет, пока не станут известны точные механизмы возникновения болезни — то есть, что именно происходит на уровне отдельных клеток, когда уничтожаются свои собственные меланоциты, и что является причиной этой «междоусобицы». В наш век постгеномных технологий [1] принято подходить к изучению генетической подоплёки заболеваний достаточно формально — генотипируя большие группы пациентов с этой болезнью и сравнивая результаты с «контрольной» группой (состоящей из здоровых людей). При этом, чтобы ничего не упустить, дефекты ищут по всему геному, сравнивая отличия в сотнях тысяч мест по всем хромосомам.
Примерный размер генома человека — три миллиарда пар оснований, однако большая часть этого материала идентична не только для любых двух людей, но и, скажем, для человека и шимпанзе (или даже мыши). Основная масса отличий кроется в так называемых однонуклеотидных заменах (или снипах — от SNP, single nucleotid polymorphism) — различиях в отдельных «буквах», составляющих «слово» (ген). При этом варианты одного гена, отличающиеся по одной (или нескольким) таким «буквам», будут называться аллельными.
Кстати, подавляющее количество снипов находится не в пределах генов, кодирующих белки (которых всего-то чуть больше 20 тысяч [2]), а в «межгенных пространствах», составляющих основную массу ДНК. Роль этой «тёмной материи» ещё совсем недавно представлялась настолько непонятной, что эти области даже называли «мусорной ДНК» [3], но на сегодняшний день накопилась уже масса свидетельств тому, что этот «балласт» на самом деле выполняет важнейшие регуляторные функции. Кстати, возможно, что именно эта ДНК играет решающую роль в эволюции организмов и определяет отличие, например, между человеком и остальными приматами [4].
Так или иначе, несмотря на медленное, но неотвратимое приближение эры «персональной геномики», когда каждый человек вместе со свидетельством о рождении будет получать и медицинскую карту с полной последовательностью своего генома [5], сейчас медицинские генетики сравнивают между собой не целые геномы, а только наборы снипов, соответствующих различиям отдельных индивидов между собой. Эта операция называется генотипированием, и может осуществляться, например, с помощью ДНК-микрочипов, способных дать информацию сразу о сотнях тысяч (до миллиона!) однонуклеотидных замен. Такие исследования пока существенно дешевле полного прочтения ДНК индивида, а большое число исследуемых снипов позволяет рассчитывать, что найденные различия укажут на место в хромосоме, предположительно связанное с тем или иным заболеванием.
В основе масштабных генетических сканирований с анализом ассоциации (или сцепления) генов (Genome-wide association/linkage study) находится статистическая процедура, определяющая значимость различия того или иного снипа между группами больных и здоровых людей. Аналогично простейшей математической статистике, происходит расчёт вероятности того, что генетическое отличие по конкретной позиции случайно, и если эта вероятность достаточно мала (например, меньше 10 −6 ), такую гипотезу отвергают. Другими словами, это будет означать, что данная замена (мутация) в геноме не случайна, то есть как-то связана с заболеванием (по наличию которого, собственно, и различаются сравниваемые группы пациентов). Когда учёные заявляют (а потом газетчики на своих длинных языках разносят), что «такой-то ген связан с развитием заболевания такого-то», в большинстве случаев речь идёт именно о таком анализе.
Но вернёмся к витилиго. Крупная международная команда учёных провела серию масштабных генетических сканирований, в результате которых были выделены несколько генов, предположительно связанных с риском развития этого заболевания [6], [7]. В одно из исследований вошли 1514 пациентов с витилиго и уже «готовые» публично доступные генотипы 2813 здоровых личностей («контроль»), а в другое — 647 больных и 1056 здоровых людей, прогенотипированных специально для этой работы. Во всех случаях генотипируемые были европейского происхождения. Для исследования некоторых нюансов в одном из исследований работа проводилась на мультиплексных семьях (в которых присутствуют несколько больных, состоящих в родстве). Для генов-«кандидатов», выбранных по данным анализа генетической ассоциации, проверяли «репликацию» ассоциации, то есть на двух независимых выборках больные/здоровые заново выясняли, сохраняется ли найденная закономерность ассоциации определённого гена с риском развития витилиго. Результаты первого исследования [6] показаны на рисунке 2.

Рисунок 2. Полногеномное сканирование с анализом ассоциации генов. На рисунке приведены данные о генотипировании 520 460 «снипов» (однонуклеотидных замен), раскрашенные по хромосоме, в которой они расположены. Ордината каждой точки соответствует отрицательному логарифму вероятности того, что отличие последовательности ДНК в этом снипе между группой больных и здоровых — случайное. Проще говоря, чем «выше» находится точка, тем больше шанс того, что соответствующая замена связана с развитием заболевания. Пунктирная линия показывает порог, «выше» которого отличия считались значимыми (P
- RERE — ген повторяющегося дипептида аргинин/глутамат (RE), участвующего в генетической регуляции и в некоторых случаях запускающего апоптоз («запрограммированную» гибель клетки);
- PTPN22 — ген лимфоидной тирозинфосфатазы, мутации в котором могут вызывать диабет первого типа, ревматоидный артрит, волчанку, диффузный токсический зоб;
- HLA-A / HLA-DRB1 — гены белков главного комплекса гистосовместимости подтипов I и II, расположенные в шестой хромосоме. Эти белки отвечают за массу иммунных функций, в частности — за презентирование антигенов Т-лимфоцитам;
- IL2RA — ген α-цепи рецептора интерлейкина 2. Этот белок уничтожается трипаносомой при болезни Шагаса, что вызывает хроническое ингибирование иммунитета;
- TYR — ген тирозиназы, отвечающей за синтез меланина. Мутация в этом гене также может привести к альбинизму.
Что интересно, подавляющее большинство генов, «уличённых» в связи с витилиго, уже замечены во взаимоотношениях с другими аутоиммунными заболеваниями (см. рисунок), и только ген тирозиназы (TYR), непосредственно участвующей в синтезе меланина, не имеет никакого отношения к иммунной системе. Мутантная форма тирозиназы (R 402 →Q 402 ) практически перестаёт делать пигмент (что и приводит к появлению белых пятен), а заодно такой её вариант становится иммуногенным для собственного организма, — именно мутантая тирозиназа является основным аутоантигеном при витилиго (и это «добивает» меланоциты). Однако оказывается, что нет худа без добра: аллельная форма тирозиназы, характерная для витилиго, оказалась исключающей по отношению к другому аллельному варианту, вызывающему меланому! (Другими словами — увеличивающийся риск витилиго, связанный с этой заменой, «автоматически» снижает вероятность возникновения опухоли.)
Второе исследование позволило добавить к уже найденным ещё пару «иммунных» генов, также связанную с развитием витилиго [7]. Ген FOXP1 кодирует транскрипционный фактор, играющий решающую роль в развитии B- и T-лимфоцитов, а также моноцитов. Ген CCR6 кодирует мембранный хемокиновый рецептор 6, распознающий воспалительный медиатор (хемокин) CCL20, выделяемый макрофагами.
Как видно из всего этого «винегрета», объяснить развитие заболевания каким-то одним фактором, по-видимому, принципиально невозможно. Кстати, по оценкам исследователей, найденные ими мутации позволяют объяснить не более 10% «генетического риска» возникновения заболевания. Развитие витилиго может начаться с разных «концов», но, с другой стороны, в будущем это позволит применять различные типы лечения и терапии, дополняющие друг друга.
«Витилиго — сложное и многофакторное заболевание, включающее не только генетику, но и многочисленные факторы среды, — говорит Маргарет «Пегги» Уоллес (Margaret Wallace), профессор молекулярной генетики и микробиологии, член Института генетики и Центра эпигенетики во Флоридском университете и одна из ведущих авторов работы. — Видимо, и для терапии существует множество возможностей. Хорошенько изучив все „тропинки“, по которым может продвигаться витилиго, мы, в конце концов, научимся пресекать путь заболевания. Кроме того, очевидно, что это — отличный вариант для персонализованной медицины будущего, когда лечение можно будет подбирать с учётом индивидуальных генетических особенностей каждого пациента» [8].
Первоначально статья опубликована в журнале «Косметика & Медицина» [9].
Что такое синдром Жильбера? Причины возникновения, диагностику и методы лечения разберем в статье доктора Васильева Романа Владимировича, гастроэнтеролога со стажем в 15 лет.
Над статьей доктора Васильева Романа Владимировича работали литературный редактор Маргарита Тихонова , научный редактор Сергей Федосов

Определение болезни. Причины заболевания
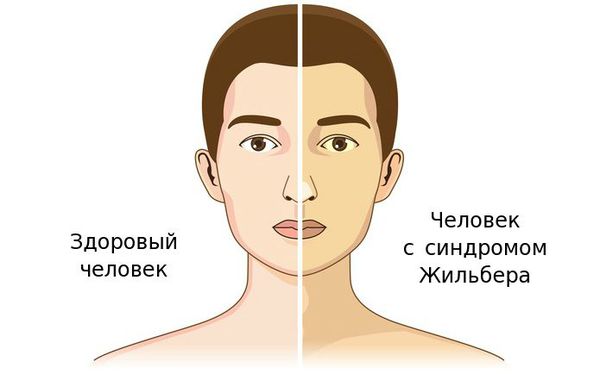
Синдром Жильбера — это генетический пигментный гепатоз с аутосомно-доминантным типом наследования, протекающий с повышением уровня неконъюгированного (свободного) билирубина, чаще проявляющееся в период полового созревания и характеризующийся доброкачественным течением [1] .
Краткое содержание статьи — в видео:
Синонимы названия болезни: простая семейная холемия, конституциональная или идиопатическая неконъюгированная гипербилирубинемия, негемолитическая семейная желтуха.
По распространённости данное заболевание встречается не менее, чем у 5 % населения, в соотношении мужчин и женщин — 4:1. Впервые заболевание описал французский терапевт Августин Жильбер в 1901 году.
Чаще синдром Жильбера проявляется в период полового созревания и характеризуется доброкачественным течением. Основным проявлением этого синдрома является желтуха.
К провоцирующим факторам проявления синдрома можно отнести:
- голодание или переедание;
- жирную пищу;
- некоторые лекарственные средства;
- алкоголь;
- инфекции (грипп, ОРЗ, вирусный гепатит);
- физические и психические перегрузки;
- травмы и оперативные вмешательства.
Причина заболевания — генетический дефект фермента УДФГТ1*1, который возникает в результате его мутации. В связи с этим дефектом функциональная активность данного фермента снижается, а внутриклеточный транспорт билирубина в клетках печени к месту соединения свободного (несвязанного) билирубина с глюкуроновой кислотой нарушается. Это и приводит к увеличению свободного билирубина.
При обнаружении схожих симптомов проконсультируйтесь у врача. Не занимайтесь самолечением - это опасно для вашего здоровья!
Симптомы синдрома Жильбера
Некоторые специалисты трактуют синдром Жильбера не как болезнь, а как физиологическую особенность организма.
До периода полового созревания данный синдром может протекать бессимптомно. Позже (после 11 лет) возникает характерная триада признаков:
- желтуха различной степени выраженности;
- ксантелазмы век (жёлтые папулы);
- периодичность появления симптомов [1] .
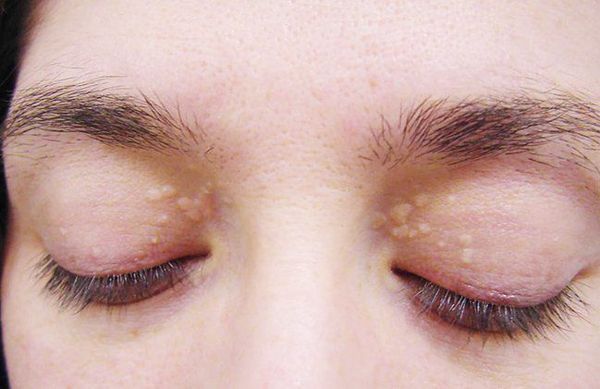
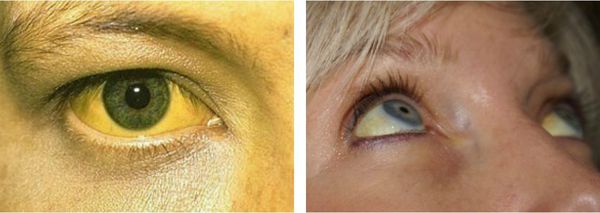
Желтуха чаще всего проявляется иктеричностью (желтушностью) склер, матовой желтушностью кожных покровов (особенно лица), иногда частичным поражением стоп, ладоней, подмышечных впадин и носогубного треугольника.
Заболевание нередко сочетается с генерализованной дисплазией (неправильным развитием) соединительной ткани.
Усиление желтухи может наблюдаться после перенесения инфекций, эмоциональной и физической нагрузки, приёма ряда лекарственных препаратов (в частности, антибиотиков), голодания и рвоты.
Клиническими проявлениями заболевания общего характера могут быть:
- слабость;
- недомогание;
- подавленность;
- плохой сон;
- снижение концентрации внимания.
В отношении ЖКТ синдром Жильбера проявляется снижением аппетита, изменением привкуса во рту (горечь, металлический привкус), реже возникает отрыжка, тяжесть в области правого подреберья, иногда наблюдается боль ноющего характера и плохая переносимость лекарственных препаратов.
При ухудшении течения синдрома Жильбера и существенном повышении токсичной (свободной) фракции билирубина может появляться скрытый гемолиз, усиливая при этом гипербилирубинемию и добавляя в клиническую картину системный зуд.
Патогенез синдрома Жильбера
В норме свободный билирубин появляется в крови преимущественно (в 80-85 % случаев) при разрушении эритроцитов, в частности комплекса ГЕМ, входящего в структуру гемоглобина. Это происходит в клетках макрофагической системы, особенно активно в селезёнке и купферовских клетках печени. Остальная часть билирубина образуется из разрушения других гемсодержащих белков (к примеру, цитохрома P-450).
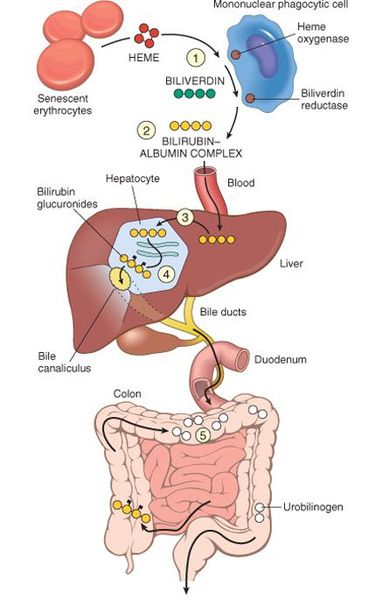
У взрослого человека в сутки образуется приблизительно от 200 мг до 350 мг свободного билирубина. Такой билирубин слаборастворим в воде, но при этом хорошо растворяется в жирах, поэтому он может взаимодействовать с фосфолипидами ("жирами") клеточных мембран, особенно головного мозга, чем можно объяснить его высокую токсичность, в частности токсичное влияние на нервную систему.
Первично после разрушения комплекса ГЕМ в плазме билирубин появляется в неконъюгированной (свободной или несвязанной) форме и транспортируется с кровью при помощи белков альбуминов. Свободный билирубин не может проникнуть через почечный барьер за счёт сцепления с белком альбумином, поэтому сохраняется в крови.
В печени несвязанный билирубин переходит на поверхность гепатоцитов. С целью снижения токсичности и выведения в клетках печени свободного билирубина при помощи фермента УДФГТ1*1 он связывается с глюкуроновой кислотой и превращается в конъюгированный (прямой или связанный) билирубин. Конъюгированный билирубин хорошо растворим в воде, он является менее токсичным для организма и в дальнейшем легко выводится через кишечник с желчью.
При синдроме Жильбера связывание свободного билирубина с глюкуроновой кислотой снижается до 30% от нормы, тогда как концентрация прямого билирубина в желчи увеличивается.
В основе синдрома Жильбера лежит генетический дефект — наличие на промонторном участке A(TA)6TAA гена, кодирующего фермент УДФГТ1*1, дополнительного динуклеотида ТА. Это становится причиной образования дефектного участка А(ТА)7ТАА. Удлинение промонторной последовательности нарушает связывание фактора транскрипции IID, в связи с чем уменьшается количество и качество синтезируемого фермента УДФГТ1, который участвует в процессе связывания свободного билирубина с глюкуроновой кислотой, преобразуя токсичный свободный билирубин в нетоксичный связанный.
Вторым механизмом развития синдрома Жильбера является нарушение захвата билирубина микросомами сосудистого полюса клетки печени и его транспорта глутатион-S-трансферазой, которая доставляет свободный билирубин к микросомам клеток печени.
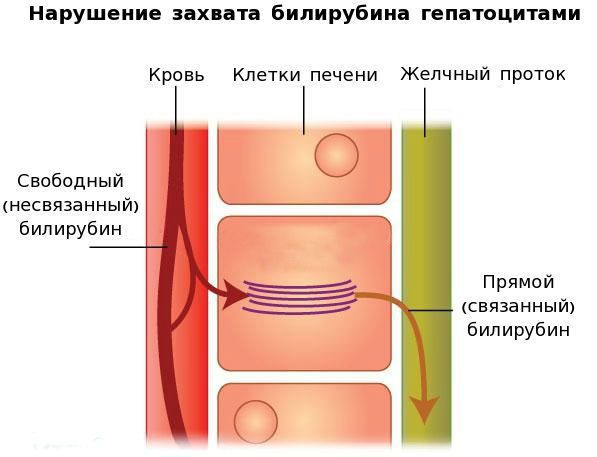
В конечном итоге вышеперечисленные патологические процессы приводят к увеличению содержания свободного (несвязанного) билирубина в плазме, что обуславливает клинические проявления заболевания [6] .
Классификация и стадии развития синдрома Жильбера
Общепринятой классификации синдрома Жильбера не существует, однако условно можно разделить генотипы синдрома по полиморфизму.
Читайте также: