
Поздняя кожная порфирия что это такое
Обновлено: 24.04.2024
Информацию из данного раздела нельзя использовать для самодиагностики и самолечения. В случае боли или иного обострения заболевания диагностические исследования должен назначать только лечащий врач. Для постановки диагноза и правильного назначения лечения следует обращаться к Вашему лечащему врачу.
Порфирия: причины появления, симптомы, диагностика и способы лечения.
Определение
Порфирии (от греч. porphyreis – пурпурный) – это ряд заболеваний обмена веществ, при которых нарушается образование гема, представляющего собой комплексное соединение порфиринов с двухвалентным железом. В результате в организме происходит накопление порфиринов (пигментов, представляющих собой производные порфина) или их предшественников. К счастью, эти патологии встречаются относительно редко ‒ не более 7-12 случаев на 100 000 человек.
Причины появления порфирии
В подавляющем большинстве случаев причиной порфирий становятся генетические мутации, обусловливающие неполноценность активности того или иного фермента, участвующего в синтезе гема. Исключением является поздняя кожная порфирия (спорадическая форма), которая развивается на фоне заболеваний печени (алкогольного гепатита, вирусного гепатита С) или длительной интоксикации тяжелыми металлами.

Наследование порфирий происходит по аутосомно-доминантному (наследуется одна нормальная и одна измененная копия гена, причем измененная копия доминирует и «подавляет» нормальную, в результате чего развивается генетическое заболевание) или аутосомно-рецессивному типу (болезнь Гюнтера). Синтез гема проходит 8 последовательных этапов, за каждый из которых отвечает конкретный фермент, кодируемый определенным геном. Соответственно, для каждой формы порфирии существует специфичный ферментативный дефект.
Наибольшее количество гемов образуется в печени и костном мозге. В печени гемы входят в состав белков, участвующих в клеточном дыхании, расщеплении токсичных свободных радикалов и «обезвреживании» различных ксенобиотиков (чужеродных химических веществ, попадающих в организм извне). В костном мозге гемы необходимы для синтеза гемоглобина.
Результатом снижения активности ферментов становится торможение синтеза гемов, что ведет к накоплению его токсичных промежуточных метаболитов.
Провоцирующими факторами развития порфириновой болезни становятся:
- избыточная инсоляция;
- недостаточное питание и скудный рацион;
- систематические стрессы;
- чрезмерное употребление алкоголя;
- вирусные и бактериальные инфекции;
- хронические интоксикации солями тяжелых металлов.
- Беременность.
В отдельных случаях манифестация патологического процесса может произойти на фоне приема некоторых лекарственных препаратов: антибиотиков, антиконвульсантов, нестероидных противовоспалительных средств, пероральных контрацептивов.
Классификация заболевания
В основу разных классификаций порфирий положены различные критерии: клиническая симптоматика, локализация нарушения метаболизма порфиринов или тканевая тропность.
Современная классификация порфирий весьма разветвленная:
I. Эритропоэтические порфирии.
- Эритропоэтическая уропорфирия (врожденная эритропоэтическая порфирия, или болезнь Гюнтера).
- Эритропоэтическая протопорфирия:
- манифестная форма;
- латентная форма.
- Эритропоэтическая копропорфирия.
- Пирролопорфирия (острая перемежающая порфирия, доминантная порфирия шведского типа):
- манифестная форма;
- латентная форма.
- Протокопропорфирия (варигатная, или смешанная порфирия, доминантная порфирия Южно-Африканского типа):
- кожная форма;
- острая форма без кожных проявлений;
- комбинированная форма с кожными и острыми проявлениями;
- латентная форма.
- Урокопропорфирия (поздняя кожная порфирия):
- манифестная приобретенная (симптоматическая) форма (развивается при интоксикациях гексахлорбензолом, опухолях печени и других патологических состояниях;
- манифестная наследственная форма (развивается у лиц с наследственной предрасположенностью);
- латентная наследственная форма (выявляется у родственников больных).
- Наследственная копропорфирия.
- Неклассифицированная печеночная порфирия.
- Гепатоэритропоэтическая порфирия.
- Неклассифицированная печеночная порфирия, протекающая с клиническим синдромом световой оспы.
- Патологические состояния, сопровождающиеся геморрагическим синдромом и нарушениями порфиринового обмена печеночного типа.
- Эритропоэтические. К ним относятся врожденная эритропоэтическая порфирия (ВЭП, болезнь Гюнтера), эритропоэтическая протопорфирия (ЭПП). Основной клинический признак – поражение участков кожи, которые подвергаются воздействию прямых солнечных лучей (фотосенсибилизация). Данные патологии являются наиболее тяжелыми и имеют самый высокий процент летальности.
- Острые печеночные. Сюда включены острая перемежающаяся порфирия (ОПП), вариегатная порфирия (ВП), наследственная копропорфирия (НКП). Острые порфирии характеризуются приступообразным течением. Преимущественно страдает нервная система. При ВП и НКП также встречаются признаки фотосенсибилизации.
- Хронические печеночные. К ним относят позднюю кожную порфирию (ПКП), которая имеет наследственную и приобретенную формы. Это наиболее благоприятный вид порфирии.
- Клинико-биохимические анализы. При болезни Гюнтера в общем и биохимическом анализах крови выявляются признаки гемолиза (пойкилоцитоз, анизоцитоз, сфероцитоз, ретикулоцитоз, повышение непрямого билирубина и сывороточного железа), увеличение печеночных трансаминаз. У больных с острыми порфириями отмечается снижение уровня глюкозы и натрия, при ПКП - увеличение сывороточного железа и ферритина. Также у 80% с ПКП выявляются положительные маркеры вируса гепатита С.
- Специфические исследования. Для диагностики острых порфирий широко используется скрининговая проба Эрлиха (при смешивании специального реактива с мочой она окрашивается в красный цвет). Для ОПП характерно повышение ДАЛК и порфобилиногена в моче, для ВП - протопорфирина в кале, для НКП – копропорфирина в кале и моче. При ПКП в моче увеличено содержание уропорфирина, в кале – копропорфирина. При ЭПП наблюдается высокая концентрация протопорфирина в эритроцитах и кале, при ВЭП - уропорфирина в моче, кале и эритроцитах. При люминесцентной микроскопии плазма дает красное флуоресцирование при ПКП и эритропоэтических порфириях.
- Острые. Для подавления образования порфобилиногена и ДАЛК применяют гем-аргинат, производные АТФ (аденил, рибоксин) и большие дозы глюкозы с дальнейшим переходом на высокоуглеводную диету. Для купирования вегетативной симптоматики используют октреотид, для ускорения восстановления миелина в нервных волокнах - витамины группы В. При менструалозависимых атаках эффективна овариосупрессивная терапия. С этой целью назначают агонисты гонадотропин-рилизинг гормона.
- Эритропоэтические. Данные порфирии очень плохо поддаются терапии. Основное лечение – это защита кожных покровов от солнечного света (окна со стеклом, не пропускающим ультрафиолет, закрытая одежда, фотозащитные кремы, прием бета-каротина). Необходимо обрабатывать эрозии антисептическими растворами для профилактики инфекционных осложнений. При выраженном гемолизе показана спленэктомия. В некоторых случаях болезни Гюнтера эффективной оказывается трансплантация костного мозга. При ЭПП дополнительно назначаются гепатопротекторы (урсодезоксихолевая кислота, адеметионин) и антицирротическая терапия.
- ПКП. С целью удаления порфиринов и избытка железа проводят плазмаферез и флеботомию (кровопускания). При противопоказаниях к данным процедурам назначаются аминохинолиновые (хлорохин) и комплексообразующие (дефероксамин) препараты. Для уменьшения всасывания порфиринов в желудочно-кишечном тракте применяют энтеросорбенты (активированный уголь). Также используются солнцезащитные кремы. При наличии гепатита С необходима противовирусная терапия интерфероном-альфа и рибавирином.
- Скрытое носительство мутантного гена. Симптоматика и биохимические изменения отсутствуют. Выявляется лишь снижение уровня эритроцитарной порфобилиногендезаминазы.
- Здоровое носительство дефектного гена. Наблюдаются минимальные биохимические изменения при отсутствии клинических проявлений.
- Латентная форма. Выявляется стертая симптоматика без типичных острых приступов болезни и выраженные нарушения порфиринового обмена.
- Манифестная форма. Отмечается яркая клиническая картина, протекающая в виде приступов. Приступы могут иметь постоянную выраженность или, наоборот, с каждым разом усиливаться. Может развиться острейшая форма (часто возникает при беременности) с грубыми неврологическими нарушениями и летальным исходом. Отмечаются выраженные биохимические отклонения порфиринового обмена.
- Исследование мочи. В общем анализе мочи выявляется повышенная относительная плотность, розовый или красно-бурый цвет мочи. Часто такую окраску моча приобретает только при длительном нахождении на свету. Существует специальный скрининговый тест с реактивом Эрлиха: при добавлении реактива к моче она в течение 5 минут становится красной. Этот анализ считается тестом первой линии, его обязательно следует проводить всем пациентам, как только у них заподозрена порфирия. При положительном результате выполняется количественное определение содержания в моче порфиринов, ДАЛК и ПБГ - при перемежающейся порфирии они повышены.
- Исследование крови. В биохимическом анализе часто обнаруживаются гипогликемия, снижение уровня натрия и осмолярности плазмы. В эритроцитах отмечается сниженная активность порфобилиногендезаминазы. При молекулярно-генетической диагностике выявляется мутация гена PBGD. Определение активности фермента и ДНК-диагностика в основном применяются при латентных и скрытых формах острой перемежающейся порфирии или при сомнительных результатах предыдущих анализов.
Симптомы порфирии
Сроки дебюта рассмотренных выше форм заболевания различны: эритропический тип проявляется в возрасте 3–5 лет, острый печеночный — в 14–16 лет, хронический печеночный — после 40 лет. После манифестации патологии пациенты сталкиваются со специфической симптоматикой.
При острых формах порфирии больные жалуются на сильные боли в животе, задержку стула, учащенное сердцебиения, повышение артериального давления, изменение цвета мочи (от розового до красно-бурого).
Тяжесть состояния пациента в основном обусловлена неврологическими симптомами – болью и снижением чувствительности по всему телу, прогрессирующей мышечной слабостью, судорожными припадками, различными психическими расстройствами (тревожностью, психомоторным возбуждением, бредом).
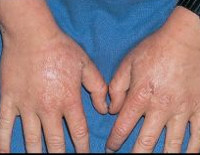
При поздней кожной форме на участках кожного покрова, подвергающихся постоянному воздействию солнечного света, формируется гиперпигментация, в результате чего кожа приобретает землистый или бронзовый оттенок.
Выраженными симптомами этой формы порфирии являются везикулезные и буллезные высыпания, которые покрыты корками, шелушение кожи, медленно заживающие эрозии, милиумы.
Кроме того, могут отмечаться явления гипертрихоза (избыточного оволосения) лобно-височной области лица, фотосенсибилизация.
При эритропоэтических порфириях наблюдаются признаки светочувствительности, причем даже более отчетливые, чем при поздней кожной порфирии. Пациент испытывает сильную боль, если на кожный покров попадают прямые солнечные лучи. Обширные эрозии оставляют после себя атрофичексие рубцы, что приводит к обезображиванию внешнего вида больного. В результате множественных атрофических рубцов на коже кистей рук развиваются контрактуры. Избыточное количество порфиринов приводит к тому, что зубная эмаль приобретает красновато-коричневый цвет (эритродонтия), моча становится красной или розовой.
Специфический признак эритропоэтической порфирии – утолщение, огрубение и уплотнение кожи периоральной и периорбитальной зон, крыльев и спинки носа, тыльной поверхности кистей.
Диагностика порфирии
При постановке диагноза учитывается наличие данного заболевания у близких родственников, возраст больного, обстоятельства возникновения симптомов (инсоляция, прием лекарств или алкоголя, голодание, инфекции, беременность).
-
Общеклинический № и биохимический анализ крови: АЛТ, АСТ, непрямой билирубин, сывороточное железо, ферритин.
Синонимы: Общий анализ крови, ОАК. Full blood count, FBC, Complete blood count (CBC) with differential white blood cell count (CBC with diff), Hemogram. Краткое описание исследования Клинический анализ крови: общий анализ, лейкоформула, СОЭ См. также: Общий анализ – см. тест № 5, Лейкоцит.
Порфирии ‒ большая группа наследственных заболеваний, характеризующихся нарушением биосинтеза гема и накоплением его токсичных метаболитов. Клинические проявления крайне разнообразны – от светочувствительности и кожных высыпаний до болей в животе, полного паралича и острых психозов. Диагностика осуществляется с помощью молекулярно-генетических тестов, специальных лабораторных методов определения порфиринов и их предшественников в моче и кале, оценки активности ферментов в крови. Лечение заключается в мероприятиях, направленных на снижение образования токсических метаболитов, их выведение из крови, проведении симптоматической терапии и хирургических вмешательств.
МКБ-10
Общие сведения
Порфирии (от греч. «porphyreis» - пурпурный) – ряд заболеваний обмена веществ, при которых нарушается образование гема, в результате чего в организме накапливаются порфирины или их токсичные предшественники. Патологии данной группы встречаются относительно редко ‒ от 7 до 12 случаев на 100 000 человек. Отдельные нозологии имеют свою эндемичность. Так, распространенность поздней кожной порфирии в странах Южной Африки составляет 1:800, острой перемежающейся порфирии в Швеции ‒ 1:1000, вариегатной порфирии в Южной Африке ‒ 1:3000. У большинства порфирий нет гендерных различий, кроме поздней кожной формы (чаще страдают мужчины) и острой интермиттирующей (чаще болеют женщины).
Причины порфирий
В подавляющем большинстве случаев причиной порфирий выступают генетические мутации, обусловливающие неполноценность активности того или иного фермента, участвующего в биосинтезе гема. Исключением является поздняя кожная порфирия (спорадическая форма), которая развивается вследствие заболеваний печени (алкогольный гепатит, вирусный гепатит С) или длительной интоксикации тяжелыми металлами. Наследование порфирий происходит по аутосомно-доминантному или аутосомно-рецессивному типу. Синтезирование гема протекает в 8 последовательных этапов, за каждый отвечает свой фермент, кодируемый определенным геном. Для каждой формы порфирии существует специфичный ферментативный дефект.
Гем представляет собой комплексное соединение порфиринов с двухвалентным железом. Наибольшее количество гема образуется в печени и костном мозге. В печени гем входит в состав белков, участвующих в клеточном дыхании, расщеплении токсичных свободных радикалов и обезвреживании различных ксенобиотиков. В костном мозге гем используется для образования гемоглобина. Результатом сниженной активности ферментов является торможение синтеза гема на определенном уровне, что ведет к накоплению его токсичных промежуточных метаболитов.
Помимо генетической мутации, для развития острых порфирий необходимо воздействие провоцирующих факторов, стимулирующих выработку порфиринов. Такими факторами являются голодание, длительная инсоляция, стрессы, алкоголь, инфекции, интоксикация тяжелыми металлами (ртуть, свинец), лекарственные средства, подвергающиеся метаболизму системой цитохрома P-450 (нестероидные противовоспалительные препараты, антибиотики, антиконвульсанты, оральные контрацептивы, седативные средства). Особую роль играют колебания женских половых гормонов во время менструаций или беременности. У женщин месячные являются наиболее частым провоцирующим фактором, а беременность ассоциируется с тяжелым течением заболевания.
Патогенез
В результате неполноценности ферментов, участвующих в образовании гема, и действия провоцирующих факторов происходит увеличение концентрации его токсичных продуктов обмена. Для хронических порфирий характерно накопление протопорфирина, копропорфирина и упопорфирина. При острых формах возрастает количество порфобилиногена и дельта-аминолевулиновой кислоты (ДАЛК).
Порфирины накапливаются в коже и под действием ультрафиолетового излучения (солнечного света) запускают процесс перекисного окисления липидов, вызывая деструкцию и гибель клеток кожи. Копропорфирин и протопорфирин усиливают пигментацию кожи и ускоряют рост волос (гипертрихоз). Плохо растворимый в воде протопорфирин откладывается в клетках печени, закупоривает портальные тракты и желчные протоки. Отложение уропорфирина в эритроцитах приводит к их ускоренному разрушению в селезенке (гемолиз). Предшественники порфиринов (ДАЛК и порфобилиноген), накапливаясь в нервной ткани, вызывают демиелинизацию и аксональную дегенерацию нервных волокон.
Классификация
В основу разных классификаций порфирий положены различные критерии: клиническая симптоматика, локализация нарушения метаболизма порфиринов или тканевая тропность. Наиболее целесообразно выделять следующие виды порфирий:
Симптомы порфирий
Спектр клинических проявлений очень широк. Порфирии могут протекать в виде острых атак или хронически. Различия наблюдаются также в возрасте дебюта заболевания. Так, эритропоэтические порфирии манифестируют уже в дошкольном детстве (3-5 лет), острые порфирии - после полового созревания (14-16 лет), а спорадическая (приобретенная) форма ПКП - после 40 лет.
При острых порфириях развиваются сильные боли в животе, задержка стула, учащение сердцебиения, повышение артериального давления, изменение цвета мочи (от розового до красно-бурого). Тяжесть состояния пациента в основном обусловлена неврологическими симптомами – болью по всему телу, снижением чувствительности, прогрессирующей мышечной слабостью, иногда достигающей полного паралича, судорожными припадками, различными психическими расстройствами (тревожность, психомоторное возбуждение, бред, галлюцинации).
При поздней кожной форме возникает гиперпигментация участков кожи, подвергающихся постоянному воздействию солнечного света (лицо, шея, ушные раковины, верхняя часть груди, кисти рук). Кожа приобретает землистый или бронзовый оттенок. Также характерны гипертрихоз лобно-височной области лица, фотосенсибилизация, проявляющаяся повышенной ранимостью кожи и образованием пузырей с жидкостным содержимым. После вскрытия пузырей формируются эрозии. На местах разрешения эрозий образуются атрофические рубцы.
При эритропоэтических порфириях наблюдаются более выраженные признаки светочувствительности, чем при ПКП (ранимость, пузыри, эрозии). При длительном нахождении на свету появляется покраснение и сильное жжение кожи. Обширные эрозии оставляют после себя грубые рубцы на лице, что приводит к обезображиванию внешнего вида больного. В результате множественных рубцов на коже кистей рук развиваются контрактуры суставов, что значительно затрудняет их движения. Моча становится красной или розовой, а зубы окрашиваются в красно-коричневый цвет (эритродонтия). Из-за увеличенной селезенки могут появиться тяжесть или ноющие боли в левом подреберье. Специфический признак ЭПП – утолщение, огрубение и уплотнение кожи вокруг рта и глаз, на крыльях и спинке носа, на тыльных поверхностях кистей.
Осложнения
Нарушения порфиринового обмена ухудшают течение сердечно-сосудистых заболеваний, неблагоприятно влияют на углеводный метаболизм и повышают риск развития сахарного диабета 2 типа. Острые формы порфирий вследствие выраженной полинейропатии осложняются параличом дыхательной мускулатуры, аспирационной пневмонией, отеком головного мозга, тромбоэмболиями, рабдомиолизом. Постоянные эрозии кожных покровов могут привести к бактериальным инфекциям. При ЭПП из-за отложения нерастворимого в воде протопорфирина может развиться цирроз печени и печеночная недостаточность.
Диагностика
При подозрении на порфирию пациента направляют к врачу-гематологу. При постановке диагноза учитывается наличие заболевания у близких родственников, возраст больного, обстоятельства возникновения симптомов (инсоляция, прием лекарств или алкоголя, голодание, инфекции, менструации, беременность). Лабораторная диагностика порфирий следующая:
Также для подтверждения диагноза проводится определение уровня ферментов цикла биосинтеза гема в эритроцитах, лимфоцитах или плазме - порфобилиногендезаминазы (ОПП), копропорфириноген-оксидазы (НКП), протопорфириноген-оксидазы (ВП), уропорфириногенсинтетазы (ВЭП), уропорфириногендекарбоксилазы (ПКП), феррохелатазы (ЭПП). Заключительным этапом диагностики является молекулярно-генетическое тестирование для выявления мутаций генов, кодирующих перечисленные выше ферменты. Данные исследования особенно эффективны для распознавания асимптомных форм порфирий.
Эритропоэтические порфирии дифференцируют с дерматологическими заболеваниями (буллезным пемфигоидом, вульгарной пузырчаткой), с гематологическими патологиями, протекающими со спленомегалией (лейкозами, лимфомами, аутоиммунными гемолитическими анемиями) с болезнями почек. ПКП дифференцируют с заболеваниями печени, гемохроматозом, надпочечниковой недостаточностью. Острые порфирии следует дифференцировать с хирургическими заболеваниями, сопровождающимися сильной болью в животе, неврологическими и психиатрическими патологиями.
Лечение порфирий
Пациентов с острыми и эритропоэтическими порфириями необходимо госпитализировать отделение гематологии. Лечение ПКП возможно как в стационаре, так и в амбулаторных условиях. На сегодняшний день не существует эффективных методов, полностью ликвидирующих нарушения обмена порфиринов. Основной упор делается на патогенетическую и симптоматическую терапию, а также на устранение провоцирующих факторов. Способы лечения зависят от вида порфирий:
Прогноз и профилактика
В большинстве случаев порфирии являются тяжелыми заболеваниями с неблагоприятным прогнозом. При эритропоэтических формах продолжительность жизни составляет около 30 лет, смерть наступает от интеркуррентных инфекций. ЭПП часто приводит к циррозу печени. При атаках острых порфирий летальный исход наблюдается в 15-20%, основная причина смерти – паралич дыхательных мышц. При ПКП прогноз благоприятный, тяжелых осложнений не происходит. Для предупреждения рецидивов рекомендуется избегать провоцирующих факторов – инфекций, голодания, стрессов, длительной инсоляции, употребления алкоголя и определенных лекарственных средств. Людям, у которых в семье есть больной порфирией, необходимо определять активность ферментов цикла синтеза гема и проводить ДНК-диагностику для выявления генетических мутаций.
1. Заболевания внутренних органов при манифестных и латентных нарушениях порфиринового обмена/ Кривошеев Б.Н. и др. – 2014.
2. Диагностика и лечение острых порфирий: Клинические рекомендации национального гематологического сообществ/ под ред. Пустовойт Я.С., Кравченко С.К., Шмакова Р.Г., Савченко В.Г. - 2018.
3. Диагностическая роль отдельных синдромов и симптомов в семиотике острых порфирий/ Пустовойт Я.С., Галстян Г.М., Савченко В.Г.//Гематология и трансфузиология. – 2014 - №59(3).
Острая перемежающаяся порфирия (ОПП) – тяжелое наследственное заболевание, характеризующееся нарушением синтеза порфиринов и накоплением их предшественников, оказывающих токсическое действие на различные органы и системы. Клинически проявляется сильной болью в животе, тошнотой, рвотой, тахикардией, гипертензией, полинейропатиями и психическими нарушениями. Диагноз ставится на основании определения повышенного содержания порфиринов и их предшественников в моче, снижения активности фермента порфобилиногендезаминазы в крови и ДНК-диагностики. Лечение заключается в подавлении образования порфиринов и симптоматической терапии.
МКБ-10

Общие сведения
Причины ОПП
В основе заболевания лежит генетически обусловленная неполноценность фермента порфобилиногендезаминазы (гидроксиметилбилансинтазы), участвующей в образовании порфиринов. Мутация расположена в гене PBGD на хромосоме 11 (локус 11q24.1-24.2). Порфирины – это большая группа органических соединений, которые являются составной частью белков, связывающих и переносящих кислород (гемоглобин, миоглобин), а также каталаз, расщепляющих перекись водорода, и цитохромов, обеспечивающих нейтрализацию ксенобиотиков (лекарства, алкоголь, яды). Синтез порфиринов осуществляется в костном мозге, печени, нейронах, эритроцитах. Дефицит порфобилиногендезаминазы при данном заболевании наблюдается главным образом в печени, а также в клетках нервной системы и эритроцитах. В результате недостаточной активности фермента происходит торможение образования порфиринов на уровне их предшественников - дельта-аминолевуленовой кислоты (ДАЛК) и порфобилиногена (ПБГ), которые являются токсичными для многих органов и систем.
Однако наличие неполноценного фермента не всегда приводит к развитию порфирии. Важную роль играют провоцирующие факторы, которые ускоряют выработку предшественников порфиринов. Такие факторы называются провоцирующими печеночными порфириногенами. Ими являются алкоголь, вирусные и бактериальные инфекции, стресс, хирургические операции, гипогликемия, голодание, колебания гормонального фона у женщин (менструация, беременность), воздействие тяжелых металлов (свинец, ртуть, висмут). Особое место отводится лекарственным препаратам, которые чаще всего вызывают манифестацию и обострение заболевания. Наиболее выраженным порфириногенным действием обладают препараты, активно метаболизирующиеся системой цитохрома P-450 - барбитураты, эстрогены, анальгин, парацетамол, гризеофульвин, сульфаниламиды, фенитоин, хлорамфеникол, карбамазепин, метилдопа.
Патогенез
В результате неполноценности фермента под действием провоцирующих факторов накапливаются промежуточные продукты метаболизма порфиринов, оказывающие свои повреждающие действия. Они блокируют натрий-калиевую АТФ-азу, тем самым изменяют метаболизм АТФ и медиаторов в нервных клетках. В результате этого развиваются демиелинизация и аксональная дегенерация нервных волокон. Эти патологические изменения больше выражены в периферической нервной системе, чем в центральной. Дистрофия в вегетативных ганглиях приводит к нарушению сосудистого тонуса и регуляции деятельности внутренних органов. Поражение абдоминальных сплетений вызывает спазм сосудов брыжейки и нарушение моторики желудка и кишечника. Вследствие ослабления активности блуждающего нерва усиливается влияние симпатической нервной системы на сердце и сосуды. Повреждение нервной системы также обусловлено возникающим дефицитом порфиринов в нейронах.
Предшественники порфиринов стимулируют выработку в гипоталамусе антидиуретического гормона. Возникает уменьшение диуреза, задержка жидкости, снижение осмолярности плазмы крови. Данное явление носит название синдром неадекватной секреции антидиуретического гормона (синдром Пархона - гидропексический синдром). Точный механизм развития этого синдрома при порфирии до сих пор неизвестен.
Классификация
В зависимости от наличия или отсутствия клинических проявлений, степени их выраженности, а также обнаружения специфических биохимических изменений в лабораторных исследованиях (повышение уровня общих порфиринов, ПБГ и ДАЛК в моче, снижение активности порфобилиногендезаминазы в эритроцитах) различают следующие формы острой перемежающейся порфирии:
Симптомы ОПП
Клинические проявления разнообразны. Чаще всего они возникают в молодом возрасте (около 20 лет). При латентной форме порфирии наблюдается следующая симптоматика – нарушение сна, склонность к артериальной гипертензии, учащенное сердцебиение, незначительные боли или неприятные ощущения в животе, снижение сухожильных рефлексов, мышечная слабость, психологические изменения личности.
При манифестной форме клиника развивается в виде острых атак. Продолжительность атаки составляет от 2 до 10 недель с рецидивами через 1-2 года. В начале приступа наиболее типичны проявления со стороны желудочно-кишечного тракта: интенсивные острые боли в животе, тошнота, рвота, вздутие, задержка стула. Боли имеют коликообразный характер, локализуются в разных отделах живота и бывают настолько сильными, что пациенты часто попадают в хирургический стационар с подозрением на острый аппендицит или перфорацию язвы желудка. Также повышается температура, увеличивается артериальное давление, вплоть до гипертонического криза, учащается сердцебиение. Характерным является розовая или красно-бурая окраска мочи.
Клиника поражения нервной системы присоединяется на 7-10 день приступа. Возникают разлитые боли по всему телу, снижается чувствительность на разных участках тела, выпадают сухожильные и кожные рефлексы. Появляются слабость и ограничение движения в конечностях (парез), более выраженные в проксимальных отделах – в плечах и бедрах. Иногда развивается полный паралич конечностей. В тяжелых случаях в патологический процесс вовлекаются черепно-мозговые нервы, что сопровождается двоением в глазах (диплопией), асимметрией лица, невнятной речью (дизартрией), нарушением глотания и поперхиванием (дисфагией).
Возможны судорожные припадки и психические нарушения – бессонница, эмоциональная лабильность, депрессивное состояние, неадекватное поведение, истерические припадки, зрительные и слуховые галлюцинации. Вследствие повышенной выработки антидиуретического гормона уменьшается мочеотделение, что приводит к водной интоксикации (гипоосмолярная гипергидратация), характеризующейся снижением аппетита, вялостью, адинамией, тремором, мышечными судорогами.
Осложнения
Наиболее тяжелые осложнения перемежающейся порфирии обусловлены полинейропатией. При параличе диафрагмы и межреберных мышц возникает острая дыхательная недостаточность, требующая проведения искусственной вентиляции легких. При слабости мышц глотки часть пищи может попасть в дыхательные пути и вызвать аспирационную пневмонию. В парализованных конечностях замедляется ток крови, что создает благоприятные условия для тромбообразования. Более редкие осложнения порфирии связаны с повышенным образованием антидиуретического гормона. Это отек головного мозга и рабдомиолиз (разрушение скелетных мышц). При рабдомиолизе из поврежденных мышечных клеток высвобождается миоглобин и калий, которые могут привести к острой почечной недостаточности и жизнеугрожающим нарушениям ритма сердца.
Диагностика
Пациентов с ОПП курирует врач-гематолог. Постановка диагноза перемежающейся порфирии представляет сложную задачу. Необходимо учитывать возраст пациента, время возникновения симптомов (например, лютеиновая фаза менструального цикла у женщин), наличие связи начала заболевания с провоцирующими факторами. Очень важно уточнять, какие лекарственные препараты принимал больной до развития приступа. Основная роль в распознавании перемежающейся порфирии отводится лабораторным методам:
Дифференциальный диагноз следует проводить с другими видами острых порфирий, демиелинизирующими заболеваниями (рассеянный склероз, синдром Гийена-Барре), отравлением свинцом, острыми абдоминальными хирургическими патологиями (аппендицит, кишечная непроходимость, холецистит, панкреатит, перфорация язвы желудка или двенадцатиперстной кишки), эпилепсией, психиатрическими болезнями. В дифференциальной диагностике принимают участие невролог, хирург, психиатр.
Лечение ОПП
Пациенты с манифестной и даже латентной формой подлежат лечению в гематологическом стационаре. При развитии выраженной неврологической симптоматики обязательна госпитализация в отделение реанимации и интенсивной терапии. Важно устранить все факторы, провоцирующие обострение заболевания. В первую очередь это касается приема лекарственных средств.
Этиотропной терапии не существует. Основная роль отводится патогенетическому лечению. Для этого применяются препараты, блокирующие образование токсичных предшественников порфиринов и, тем самым, уменьшающие их патологическое действие. К ним относятся большие дозы глюкозы, гема аргинат, сандостатин, аденил-5-монофосфат. Для ускорения регенерации миелиновой оболочки в нервных волокнах назначаются витамины группы В, для профилактики тромбообразования – антикоагулянты. Также используются антигипертензивные, анальгетические, противорвотные, слабительные, седативные препараты.
Если атаки порфирии являются менструалозависимыми и возникают часто (2-3 раза в год), необходимо подавление овуляции. С этой целью применяют агонисты гонадотропин-рилизинг гормона (Гозерелин). Беременность является неблагоприятным фактором и ассоциирована с молниеносным течением перемежающейся порфирии, высокой частотой летальных исходов. При развитии приступа в I и II триместре рекомендуется прерывание беременности, в III триместре проводится экстренное оперативное родоразрешение.
Прогноз и профилактика
Острая перемежающаяся порфирия - это тяжелое заболевание с неблагоприятным прогнозом и достаточно высоким уровнем летальности (15-20%). Самая частая причина смерти – паралич дыхательной мускулатуры вследствие полинейропатии. Очень важно своевременно диагностировать болезнь и начать специфическую терапию. Профилактика заключается в соблюдении высокоуглеводной диеты и избегании всех провоцирующих факторов, которые могут вызвать обострение – стрессов, инфекций, голодания, приема лекарственных средств и алкоголя. При наличии детей у пациентки с порфирией от новой беременности лучше отказаться. Всем родственникам больного порфирией для выявления скрытых или латентных форм заболевания необходимо проводить молекулярно-генетическую диагностику, определять уровень эритроцитарной порфобилиногендезаминазы и количество порфиринов в моче.
1. Диагностика и лечение острых порфирий. Клинические рекомендации национального гематологического сообщества/ под ред. Я.С. Пустовойт, С.К. Кравченко, Р.Г. Шмакова, В.Г. Савченко. - 2018.
2. Лабораторная диагностика острой перемежающейся порфирии/ Карпова И.В., Сурин В.Л., Тагиев А.Ф., Пивник А.В.// Проблемы гематологии и переливания крови. - 1998 - №1.
3. Острая порфирия с полиневропатией и положительным эффектом лечения глюкозой/ Котов С.В., Сидорова О.П.// Медицинская генетика. – 2016 - №7.
4. Заболевания внутренних органов при манифестных и латентных нарушениях порфиринового обмена: Моногр./Б.Н.Кривошеев и др.
П оздняя кожная порфирия – обменное заболевание, обусловленное снижением активности печеночной уропорфириноген-декарбоксилазы (УПГД), характеризующееся кожными проявлениями и поражением печени. Частые гистологическиу изменения в печени (стеатоз, сидероз, хронический гепатит, цирроз, гепатокарцинома) у больных поздней кожной порфирией требуют изучения с точки зрения инфицирования вирусами гепатита В и С, широко распространенного на юге Европы. Пока неясно, выявляет ли вирусная инфекция латентный дефицит энзима или непосредственно вызывает недостаточную активность УПГД. В обзорной статье M. Roux и соавт. обсуждаются различные современные патофизиологические гипотезы.
Клинически поздняя кожная порфирия проявляется высокой фотосенсибилизацией с буллезными поражениями на открытых местах или другими признаками повышенной "ранимости" кожи, склеродермиформными изменениями. Гепатомегалия определяется у 46% пациентов в начале заболевания и у 89% через 5 лет. Содержание уропорфиринов в моче и копропорфиринов в моче и фекалиях повышено. Часто обнаруживается повышенная активность трансаминаз (50% случаев), щелочной фосфатазы, сывороточного железа и ферритина. Эта перегрузка железом ингибирует активность УПГД. С высокой – до 95% случаев – частотой выявляются гистологические изменения в печени. Поражение паренхимы печени связывают либо с антигенной ролью порфиринов и образованием антигепатоцитных антител, либо с прямым цитотоксическим или канцерогенным действием порфиринов. Определенную роль приписывают хроническому алкоголизму, выявляемому в 1/3 случаев. Основой терапии являются препараты, уменьшающие перегрузку железом, и кровопускания. Это позволяет добиться исчезновения буллезных поражений в течение 2 – 3 мес у всех больных. Исследование активности эритроцитарной УПГД у членов семьи больного может способствовать проведению ранней превентивной терапии.
Cочетание порфирии и доброкачественных опухолей печени встречается редко; напротив, частота гепатокарциномы у больных порфирией в 2 – 3 раза выше, чем у населения в целом. Значимыми факторами риска развития гепатокарциномы на фоне порфирии являются длительность течения болезни (от 11 до 23 лет), мужской пол, возраст старше 51 года, наличие цирроза и/или хронического активного гепатита.
Частота выявления антител к вирусу гепатита С (HCV) у больных порфирией варьирует от 53 до 82%, причем 2/3 пациентов HCV-положительны в момент постановки диагноза. В 65 – 100% случаев HCV-инфекция является активной, что подтверждается полимеразной цепной реакцией. 30% HCV-положительных больных имеют одновременно маркеры вирусов С и В (HBV). Mаркеры HBV определяются у больных порфирией в 40 – 70% случаев. Роль HCV в генезе поражения печени при поздней кожной порфирии представляется большей, чем роль НVB, в связи с высокой частотой его выявления в случаях тяжелого повреждения печени и низкой частотой выявления HBsAg.
В настоящее время предложены две основные гипотезы патофизиологии поздней кожной порфирии.
Первая рассматривает эту патологию как нарушение обмена веществ, приводящее к поражению печени из-за гиперпродукции порфиринов; демаскировать латентный дефицит УПГД и вызвать проявление заболевания может множество факторов (токсины, антигены, канцерогены). Инфекция HBV и HCV, легко возникающая на фоне иммуносупрессии, вызванной дефицитом УПГД, может способствовать клиническому проявлению этого дефицита.
Вторая гипотеза исходит из первичности повреждения печени, обусловливающего ферментную недостаточность; HBV и HCV могут играть при этом непрямую патогенетическую роль.
Авторы считают, что, поскольку поздняя кожная порфирия представляет собой заболевание печени с кожными проявлениями, все такие пациенты должны быть обследованы на инфицирование HBV и HCV серологически и с определением репликации вирусов, у них необходимо определять уровень a-фетопротеина, проводить эхографию и биопсию печени. Пациенты должны обследоваться каждые 6 мес даже при отсутствии прогрессирования кожных проявлений. Пункционную биопсию следует повторять в зависимости от исходной картины и проводимой терапии. Таким образом возможно снизить летальность от гепатокарциномы на фоне поздней кожной порфирии.
Roux М, Grange С, Vital Durand D, Levrat R. Porphyrie cutanee tardive et infection par les virus de l’hepatite B et C. Presse Med 1996;25:1589–91.
Контент доступен под лицензией Creative Commons «Attribution» («Атрибуция») 4.0 Всемирная.
Авторы: Олисова О.Ю. 1 , Теплюк Н.П. 2 , Грабовская О.В. 2 , Плиева Л.Р. 2 , Орлова Е.В. 2 , Вертиева Е.Ю. 1
1 ФГАОУ ВО Первый МГМУ им. И.М. Сеченова Минздрава России (Сеченовский Университет), Москва
2 ГБОУ ВПО «Первый МГМУ им. И.М. Сеченова» МЗ РФ, Москва
Термин «паранеопластический синдром» (ПНС) в дословном переводе означает синдром, сопутствующий злокачественной опухоли. Однако, согласно данным литературы, ПНС не сопутствует опухоли, а непосредственно вызван ею. В ряде случаев ПНС может являться первым клиническим проявлением злокачественной опухоли. Иногда его характер позволяет заподозрить локализацию опухоли, т. к. определенный тип злокачественных опухолей ассоциируется со специфическим поражением кожи. Группа кератодермий преимущественно встречается у пожилых людей, и в ней существует ряд нозологий, которые рассматривают как ПНС. Среди паранеопластических заболеваний кожи выделяют кератодермии паранеопластического генеза. В статье представлены современные сведения об эпидемиологии, этиологии, патогенезе данной патологии; дана классификация кератодермий паранеопластического генеза. Описаны клинические проявления наиболее часто встречающихся паранеопластических дерматозов. Рассматриваются вопросы дифференциальной диагностики. Представлено описание больной с синдромом Лезера – Трела при умеренно дифференцированной аденокарциноме сигмовидной кишки. Онкологический диагноз был выявлен в процессе онкопоиска, проведенного в связи с дерматологическим заболеваниям.
Ключевые слова: кератодермии паранеопластического генеза, синдром Лезера – Трела, умеренно дифференцированная аденокарцинома сигмовидной кишки.
Для цитирования: Олисова О.Ю., Теплюк Н.П., Грабовская О.В. и др. Кератодермии как паранеопластический синдром // РМЖ. Дерматология. 2016. № 10. С. 654–656.
Для цитирования: Олисова О.Ю., Теплюк Н.П., Грабовская О.В. и др. Кератодермии как паранеопластический синдром. РМЖ. 2016;10:654-656.
Keratodermas as a paraneoplastic syndrome
Olisova O.Yu., Teplyuk N.P., Grabovskaya O.V., Plieva L.R., Orlova E.V., Vertieva E.Yu.
I.M. Sechenov First Moscow State Medical University, Moscow, Russia
To translate literally, paraneoplastic syndrome is a syndrome that accompanies malignant tumor. However, published data suggest that paraneoplastic syndrome is directly induced by the tumor but not accompanies it. In a number of cases, paraneoplastic syndrome can be the first clinical manifestation of the tumor. Sometimes, the nature of paraneoplastic syndrome may suggest the localization of the tumor since certain types of malignant tumors are associated with specific skin lesions. Keratodermas commonly occur in elderly patients. Some of these conditions are considered as paraneoplastic syndromes. Paraneoplastic keratodermas belong to a group of paraneoplastic skin disorders. The paper reviews current data on epidemiology, etiology, pathogenesis, and classification of these conditions. Clinical manifestations of common paraneoplastic dermatoses as well as differential diagnostic aspects are discussed. Clinical case describes a woman with Leser-Trelat sign and moderately differentiated adenocarcinoma of the sigmoid colon. The diagnosis of the malignant tumor was provided by diagnostic tests which were performed to identify the nature of skin lesions.
Key words: paraneoplastic keratoderma, sign of Leser-Trelat, moderately differentiated adenocarcinoma of the sigmoid colon.
For citation: Olisova O.Yu., Teplyuk N.P., Grabovskaya O.V. et al. Keratodermas as a paraneoplastic syndrome // RMJ. Dermatology. 2016. № 10. P. 654–656.
В статье рассмотрена проблема кератодермии, как паранеопластического синдрома
Кератодермии паранеопластического генеза отражают основной клинический признак данной группы заболеваний, а именно гиперкератоз, развивающийся при онкологическом поражении внутренних органов.
Акрокератоз псориазиформный Базекса развивается преимущественно у мужчин белой расы старше 40 лет [1, 2].
Приобретенная кератодермия ладоней и подошв (кератодермия бородавчатая узелковая Лорта – Жакоба) наблюдается редко, в доступной литературе эпидемиологические данные отсутствуют.
Синдром Лезера – Трела встречается одинаково часто у мужчин и женщин. Связь с определенной расой не выявлена.
Согласно классификации В.П. Адаскевича, С.М. Хассуаны (2002), к кератодермиям паранеопластического генеза относят [3]:
- облигатные – акрокератоз псориазиформный Базекса;
- факультативные – приобретенная кератодермия ладоней и подошв (кератодермия бородавчатая узелковая Лорта – Жакоба), синдром Лезера – Трела.
Клаус Вольф и соавт. (2012) в группе паранеопластических дерматозов выделяют гиперкератозные заболевания, включающие черный акантоз, приобретенный ихтиоз, синдром рубцевания ладоней, синдром Лезера – Трела, синдром Базекса (табл. 1) [4, 5].
Точные данные об этиопатогенезе отсутствуют. Определенный вклад в развитие заболеваний вносит нарушение гомеостаза фактора роста. При этом выявляется повышение уровней EGF и TGF-α в моче. Обращает на себя внимание снижение уровня фактора роста после удаления опухолей. Кроме того, отмечаются нарушения в экстрацеллюлярном матриксе. Предполагается первичное провоцирование гиперпролиферативного состояния посредством экстрацеллюлярного матрикса [4].
Акрокератоз псориазиформный Базекса характеризуется эритематозно-сквамозными очагами поражения акральной локализации (кисти и стопы). Развивается преимущественно у мужчин старше 40 лет. Клиническим проявлениям заболевания обычно предшествует (на месяцы или даже годы вперед) рак предстательной железы (с метастазами в лимфатические узлы шеи и средостения), верхних дыхательных путей, верхних отделов желудочно-кишечного тракта (ЖКТ), полости рта, языка, губ. Синдром наследуется по доминантному и аутосомно-доминантному типу.
Кожные изменения при акрокератозе Базекса развиваются постепенно. Первоначально появляется застойная эритема с фиолетовым оттенком и незначительным шелушением на коже носа, по краям ушных раковин, на кончиках кистей (ладони), стоп (подошвы). Сыпь располагается симметрично. Постепенно она становится генерализованной. Возникают дистрофия ногтей, паронихии. Изменения кожи лица могут носить экзематозный характер или напоминать красную волчанку, в то время как акральные участки поражения (на кистях и стопах) напоминают псориаз. В ряде случаев вначале псориазиформные очаги располагаются на тыле кистей и стоп, а позже – в области ладоней, подошв, коленных и локтевых суставов, спинке носа, завитках ушных раковин. При синдроме Базекса также нередки гипотрихоз, фолликулярная атрофия, невусы, базалиомы, милиумы, мелкие пигментные пятна, изменения ногтевых пластинок.
Гистологические изменения при акрокератозе Базекса не имеют диагностического значения и проявляются гиперкератозом, паракератозом, очагами спонгиоза и смешанным воспалительным дермальным инфильтратом. Дифференциальный диагноз проводят с себорейной экземой, аллергическим дерматитом, болезнью и синдромом Рейтера, псориазом, красной волчанкой.
Приобретенную кератодермию ладоней и подошв, а также кератодермию бородавчатую узелковую Лорта – Жакоба (КБУ) относят к факультативным паранеопластическим дерматозам [3]. КБУ описана у пациентов со злокачественной патологией печени и раком предстательной железы.
КБУ (син.: порокератоз ладонно-подошвенный папилломатозный Манту) развивается в основном у пожилых людей. При этом образуются безболезненные папилломатозные полупросвечивающие разрастания типа роговых «жемчужин» на ладонях и подошвах, в центре которых имеются точечные углубления. Развивается заболевание медленно, иногда регистрируют спонтанное излечение без рубцов. Клинически КБУ очень похожа на кератодермию Бушке – Фишера [6].
Также к факультативным паранеоплазиям относят синдром Лезера – Трела. Данный синдром характеризуется внезапным появлением себорейных кератом, быстрым увеличением их количества и размера, что в 70% случаев сочетается с онкологической патологией: раком предстательной железы, желудка, бронхов, матки, молочных желез, легкого, а также со злокачественными лимфомами. Высыпания, клинически и гистологически идентичные сенильному себорейному кератозу, обычно локализуются на спине, груди, конечностях. Клиническая картина может быть пестрой из-за вкраплений актинического кератоза, лентигинозных пятен, гемангиом [3].
Клинический случай
Больная К., 72 лет, жительница Москвы, обратилась в амбулаторное отделение клиники кожных и венерических болезней им. В.А. Рахманова.
Жалобы при поступлении на высыпания, локализованные на коже туловища, верхних конечностей, бедер. Субъективно: умеренный зуд.
Анамнез заболевания. Считает себя больной в течение 1 года, когда отметила появление сыпи под молочными железами и в области грудной клетки. Лечилась амбулаторно, проводимого лечения пациентка не помнит (медицинская документация отсутствует). Около 4-х нед. назад отметила усиление яркости окраски очагов, в связи с чем применяла антигистаминные препараты, взбалтываемую взвесь оксида цинка, топические стероиды – без эффекта. В процессе лечения отметила увеличение количества высыпаний и усиление зуда. По словам пациентки, за 3 нед. до начала прогрессирования процесса значительно увеличилось количество кератом на коже туловища и под молочными железами, изменился их характер – они стали мягкими, рыхлыми, появился неприятный запах. Обратилась в амбулаторное отделение клиники кожных и венерических болезней им. В.А. Рахманова, поступила в кожно-венерологический диспансер для уточнения диагноза и подбора адекватной терапии.
Сопутствующие заболевания: сахарный диабет 2-го типа, гипертоническая болезнь II степени, глаукома, артроз коленного сустава.
Кожный статус. Поражение кожи подостровоспалительного характера. Сыпь локализуется на коже живота, спины, под молочными железами, на верхних конечностях. На коже живота, под молочными железами визуализируются множественные себорейные кератомы до 2 см в диаметре, темно-коричневого цвета, покрытые плотными корками (рис. 1). На коже верхних конечностей, спины отмечаются эритематозно-сквамозные очаги до 10–15 см в диаметре, покрытые наслоением мелкопластинчатых чешуек. Кожа вне очагов поражения бледно-розового цвета. Волосы и ногти не поражены. Лимфатические узлы не увеличены. Тургор и эластичность соответствуют возрасту. Субъективно: интенсивный зуд.

При дерматоскопии выявлены типичные признаки кератом с крупными гиперпигментированными комедоноподобными отверстиями.
На основании клинических изменений поставлен диагноз: Себорейный кератоз. Синдром Лезера – Трела. Начато обследование больной по программе онкопоиска.
Результаты обследования. Общий анализ крови: тромбоциты – 147,8*109/л, остальные показатели в норме. Общий анализ мочи в норме. В биохимическом анализе крови – АЛТ 61 Ед/л, остальные показатели в норме.
Эзофагогастродуоденоскопия: смешанный гастрит.
УЗИ брюшной полости: диффузные изменения печени и поджелудочной железы.
Во время обследования проводилось лечение: клемастин 2,0 мл в/м н/н (10 дней), лоратадин 1 таблетка 10 мг 1 р/день (13 дней), флуконазол 1 таблетка 50 мг 1 р/день (12 дней), метформин 850 мг/сут (13 дней). Местно: мазь бетаметазон + гентамицин + клотримазол, мазь бетаметазон + салициловая кислота, порошок клотримазол, мазь эргокальциферол + токоферола ацетат + ретинола пальмитат.
Лечение перенесла хорошо, без побочных и нежелательных явлений. Со стороны кожного процесса отмечается положительная динамика в виде уменьшения яркости окраски очагов поражения на коже верхних конечностей на 70–80%. Прогрессирование себорейного кератоза не отмечается.
Рекомендовано:
1. Наблюдение у дерматолога в КВД по месту жительства.
2. Продолжить онкопоиск: маммография, колоноскопия, УЗИ малого таза в поликлинике по месту жительства.
При колоноскопии в сигмовидной кишке было обнаружено опухолевидное образование, диаметром около 3 см, красного цвета, с бугристой поверхностью, неправильной формы. При гистологическом исследовании данной экзофитно растущей опухоли были обнаружены признаки умеренно дифференцированной аденокарциномы. Больная направлена на дообследование и лечение к онкологу.
Таким образом, синдром Лезера – Трела является факультативным паранеопластическим заболеванием. При быстром возникновении множественных кератом требуется проведение онкопоиска у больных. При этом особое внимание следует обращать на состояние ЖКТ, а также исключать лимфопролиферативные заболевания. Кроме того, необходимо учитывать, что упорное течение, торпидность к проводимой терапии при паранеопластических кератодермиях отражают прогрессирование злокачественного процесса, а своевременно начатое лечение способствует положительной динамике в течение паранеопластического дерматоза.
1. Jessica G. Zarzour, Satinder Singh, Aleodor Andea, Jennifer A. CafardiAcrokeratosis paraneoplastica (Bazex syndrome): Report of a case associated with small cell lung carcinoma and review of the literature // Case Rep. J Radiol. 2011. Vol. 5(7). Р. 1–6.
2. Aline Hempen, Eleftherios P. Samartzis, Jivko Kamarachev, Daniel Fink, Konstantin J. Dedes. Acrokeratosis paraneoplastica in serous ovarian carcinoma: case report // BMC Cancer. 2015. Vol. 15. Р. 507.
3. Адаскевич В.П., Хассуана С.М. Облигатные паранеопластические дерматозы. Классификация паранеопластических дерматозов. Витебск: Витебский ГМУ. Медицинская панорама. 2002. Вып. 1 [Adaskevich V.P., Hassuana S.M. Obligatnye paraneoplasticheskie dermatozy. Klassifikacija paraneoplasticheskih dermatozov. Vitebsk: Vitebskij GMU. Medicinskaja panorama. 2002. Vyp. 1 (in Russian)].
4. Клаус Вольф, Лоуэлл А. Голдсмит, Стивен И. Кац, Барбара А. Джилкрест, Эми С. Паллер, Дэвид Дж. Леффель. Фицпатрик в клинической дерматологии. М.: изд-во Панфилова, БИНОМ, 2012. Т. 2. С. 1154–1155; 1630–1647 [Klaus Vol'f, Loujell A. Goldsmit, Stiven I. Kac, Barbara A. Dzhilkrest, Jemi S. Paller, Djevid Dzh. Leffel'. Ficpatrik v klinicheskoj dermatologii. M.: izd-vo Panfilova, BINOM, 2012. T. 2. S. 1154–1155; 1630–1647 (in Russian)].
5. Молочков В.А. Шабалин В.Н., Кряжева С.С., Романенко Г.Ф. // Руководство по геронтологической дерматологии. М., 2004. C. 193 [Molochkov V.A. Shabalin V.N., Krjazheva S.S., Romanenko G.F. // Rukovodstvo po gerontologicheskoj dermatologii. M., 2004. C. 193 (in Russian)].
6. Елькин В.Д., Митрюковский Л.С., Седова Т.Г. Избранная дерматология. Редкие дерматозы и дерматологические синдромы. Пермь, 2004. С. 506 [El'kin V.D., Mitrjukovskij L.S., Sedova T.G. Izbrannaja dermatologija. Redkie dermatozy i dermatologicheskie sindromy. Perm', 2004. S. 506 (in Russian)].
Контент доступен под лицензией Creative Commons «Attribution» («Атрибуция») 4.0 Всемирная.
Читайте также: