
Перечислите стадии поражения кожи при ювенильной склеродермии
Обновлено: 18.04.2024
Клиника склеродермии у детей. Варианты течения
Синдром Рейно (спазм артерий пальцев) может проявляться много месяцев и даже лет до обширных поражений кожи и внутренних органов. На холоде происходит спазм артерий пальцев рук и ног, а иногда ушей и кончика носа. Синдром Рейно протекает в три стадии: бледность, цианоз и эритема. Для диагностики этого нарушения достаточно двух из них. Спазмы продолжаются минуты или часы.
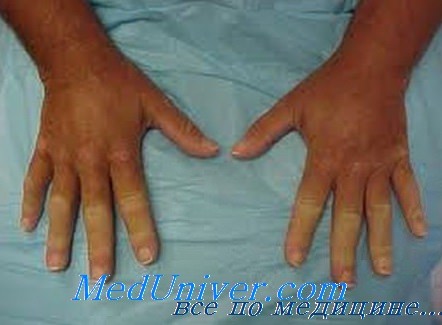
Системная склеродермия. Развитию хронического фиброза часто предшествует стадия отека, которая может длиться несколько месяцев. Вначале набухают пальцы и тыльные поверхности кистей рук, иногда и лицо. Спадая, отеки оставляют после себя натянутую кожу. Отек кистей обычно распространяется на предплечья. Атрофия подкожной клетчатки на лице приводит к сужению ротового отверстия с уменьшением расстояния между верхними и нижними зубами, даже когда рот широко открыт.
Изъязвление кожи в местах давления на нее (например, на локтях) иногда сопровождается обызвествлением подкожной клетчатки. Позднее атрофированная кожа приобретает лоснящийся вид. При тяжелом синдроме Рейно на кончиках пальцев могут появляться язвы. Дистальные фаланги подвергаются акроостеолизу. Пальцы с натянутой кожей заостряются (склеродактилия), и в конце концов развиваются вторичные сгибательные контрактуры, резко ограничивающие подвижность пальцев. Позднее из-за слабости и атрофии мышц могут развиться сгибательные контрактуры локтевых, тазобедренных и коленных суставов.
К другим последствиям хронической склеродермии относится истончение эпидермиса, облысение и снижение потоотделения. Гиперпигментированные пятна в окружении атрофических обесцвеченных участков придают некоторым повреждениям кожи вид соли с перцем. С годами возможна нормализация структуры одних пораженных участков кожи с одновременным фиброзом других.
В легких в процесс вовлекаются как артерии, так и интерстициальная ткань, что может закончиться одышкой и правожелудочковой недостаточностью. На ранних стадиях болезни рентгенография грудной клетки не всегда выявляет нарушения, их удается обнаружить только при исследовании легочной функции (определения диффузионной способности легких для СО) или с помощью КТ высокого разрешения.
Склеротические изменения могут развиваться и в других органах. Поражение почечных артерий приводит к хронической гипертонии или эпизодическим резким подъемам АД. Фиброз стенок пищевода сопровождается его расширением и дисфагией. Расширение петель кишечника может нарушать процессы всасывания и общее развитие ребенка. При фиброзе сердца наблюдаются аритмии, гипертрофия желудочков и снижение насосной функции органа.
Фиброз может распространяться только на кожу дистальных отделов конечностей, лица и шеи. На кончиках пальцев, лице, груди и внутренней поверхности губ появляются телеангиэктазии. Синдром CREST включает следующие признаки: обызвествление (кальциноз), синдром Рейно, нарушение перистальтики пищевода, склеродактилию и телеангиэктазию. У некоторых больных с этим синдромом развивается тяжелая легочная гипертензия.
Ограниченная и лентовидная склеродермия. При локализованной склеродермии обычно поражается только кожа; системный склероз развивается редко. Кольцевидная, или ограниченная, склеродермия у детей характеризуется отдельными пятнами, чаще всего на лице. Первые воспалительные проявления сменяются плотными депигментированными и атрофическими очагами. При лентовидной склеродермии фиброз захватывает кожу конечностей, иногда по всей их длине, и может распространяться на мышцы. В некоторых случаях происходит полная потеря мышечной ткани между кожей и костью.
Одна нога становится короче другой, развиваются сгибательные контрактуры суставов, а также деформации лица и лобно-теменной области, что может потребовать оперативного вмешательства.
Очаговая склеродермия – это хроническое заболевание соединительной ткани, характеризующееся преимущественным поражением кожных покровов. Клинически проявляется уплотнением (индурацией) различных участков кожи с последующей атрофией и изменением пигментации, образованием контрактур. Диагноз ставится на основании симптоматики, обнаружения в крови антинуклеарного фактора и антицентромерных антител. В сомнительных случаях проводится гистологическое исследование кожи. Лечение заключается в применении глюкокортикостероидов, иммунодепрессантов, антифиброзных средств, блокаторов кальциевых каналов и проведении ПУВА-терапии. В ряде случаев выполняются хирургические операции.
МКБ-10
Общие сведения
Очаговая (локализованная, ограниченная) склеродермия – хроническое аутоиммунное заболевание из группы диффузных болезней соединительной ткани. Патология встречается повсеместно, распространенность составляет от 0,3 до 3 случаев на 100 000 человек. Чаще страдают женщины европеоидной расы. Возраст манифестации очаговой склеродермии зависит от формы. Бляшечная склеродермия чаще встречается у взрослых (30-40 лет), линейная - у детей от 2 до 14 лет, склероатрофический лихен – у женщин старше 50 лет. При локализованной форме, в отличие от системной, поражение внутренних органов в большинстве случаев либо минимально, либо отсутствует. Имеется ассоциация склеродермии с патологиями щитовидной железы (тиреоидитом Хашимото, болезнью де Кервена).
Причины
Точная причина заболевания неизвестна. Предполагается этиологическая роль бактерии Borrelia burgdorferi, вызывающей лайм-боррелиоз, однако убедительных данных за эту теорию на сегодняшний день нет. В развитии склеродермии важную роль играет наследственная предрасположенность. Были выявлены более частые случаи очаговой склеродермии среди близких родственников. При проведении генетических исследований обнаружена взаимосвязь между определенными генами гистосовместимости (HLA – DR1, DR4) и локализованной формой заболевания. Провоцирующими факторами, способствующими возникновению склеродермии, являются переохлаждения, травмы, постоянные вибрационные воздействия на кожу, прием лекарственных препаратов (блеомицина). Триггерными эффектами также обладают различные химические соединения (хлорвинил, кремний, нефтепродукты, сицилий, эпоксидная смола, пестициды, органические растворители).
Патогенез
Выделяют три основных патогенетических механизма склеродермии – фиброз (разрастание соединительной ткани), аутоиммунное повреждение и сосудистые нарушения. Иммунная аутоагрессия заключается в выработке лимфоцитами антител к соединительной ткани и ее компонентам. Также лимфоциты синтезируют интерлейкины, которые стимулируют пролиферацию фибробластов, гладкомышечных клеток и образование коллагена. Разрастающаяся при этом соединительная ткань замещает нормально функционирующую ткань. В результате повреждения эндотелия сосудов антителами и пролиферирующими гладкомышечными клетками снижается уровень простациклина (вещества, обладающего антиагрегантными и вазодилатирующими свойствами). Это приводит к спазму микрососудов, повышению адгезии и агрегации форменных элементов крови, внутрисосудистой коагуляции и микротромбозу.
Классификация
Очаговая склеродермия подразделяется на множество форм. Наиболее распространенными являются бляшечная и линейная. У ряда пациентов могут наблюдаться одновременно несколько вариантов заболевания. Существует целый ряд классификаций, но наиболее оптимальной и широко используемой считается классификация клиники Мэйо, включающей следующие разновидности очаговой склеродермии:
- Бляшечная. Данная форма в свою очередь подразделяется на поверхностную (морфеа) и узловатую (келоидоподобную). Характерны типичные участки уплотнения кожи с атрофией и нарушением пигментации.
- Линейная. К ней относятся полосовидная, саблевидная формы, а также прогрессирующая гемиатрофия лица Парри-Ромберга. Очаги располагаются в виде линий по ходу сосудисто-нервного пучка.
- Генерализованная (многоочаговая). Проявляется сочетанием бляшечного и линейного вариантов. Очаги распространены по всему телу.
- Буллезная. При данной разновидности на коже возникают пузыри с жидкостным содержимым, оставляющие после себя эрозии.
- Пансклеротическая инвалидизирующая. Наиболее неблагоприятная форма очаговой склеродермии. Характеризуется тяжелым, прогрессирующим течением, плохо поддается лечению. Поражаются все слои кожи и ткани, лежащие под ней. Развиваются грубые контрактуры суставов и длительно незаживающие язвы на коже.
- Склероатрофический лихен Цумбуша (болезнь белых пятен). Характерно образование пятен белого цвета, сопровождающихся нестерпимым зудом. Преимущественная локализация пятен – половые органы.
Симптомы
Для клинической картины типично образование на коже очагов, которые проходят три последовательных стадий развития – отек, индурацию (уплотнение) и атрофию. В начале заболевания на коже конечностей, шеи или туловища появляются пятна сиреневого или лилового цвета, имеющие нечеткие края. Размер пятен может сильно варьировать – от просяного зерна до размеров ладони и больше. На этом этапе пациент не испытывает каких-либо неприятных ощущений или боли. Затем пятна начинают отекать, кожа в центре очага уплотняется, становится блестящей, приобретает цвет слоновой кости. Пациент начинает ощущать зуд, покалывания, стянутость кожи, болезненность. Далее наступает стадия атрофии. Кожа в очагах истончается, прекращается рост волос, нарушается потоотделение, возникает стойкая дисхромия (гипер- или депигментация) и телеангиэктазии. Иногда развивается атрофодермия (участки западения кожи).
При линейной склеродермии очаги расположены по ходу нервов и сосудов. В случае локализации на коже лица очаги по внешнему виду напоминают рубец от удара саблей (саблевидная форма). Прогрессирующая гемиатрофия представляет собой глубокий процесс с поражением всех тканей половины лица - кожи, подкожной клетчатки, мышц и костей лицевого скелета, что приводит к выраженной деформации лица, обезображивающей внешний вид пациента. Также происходит атрофия половины языка и снижение вкусовой чувствительности.
Из внекожных признаков очаговой склеродермии стоит отметить офтальмологические и неврологические проявления при гемиатрофии Парри-Ромберга. Они включают выпадение ресниц и бровей на стороне поражения, западение глазного яблока из-за атрофии глазных мышц и орбитальной клетчатки, нейропаралитический кератит, головокружения, когнитивные нарушения, мигренозные головные боли, эпилептические припадки. Также возможно развитие феномена Рейно. Симптомы синдрома Рейно следующие – стадийное изменение окраски кожи пальцев рук вследствие вазоспазма и последующей гиперемии (бледность, цианоз, покраснение), сопровождающееся онемением, болью и покалыванием в пальцах рук. Остальные экстрадермальные проявления, характерные для системной склеродермии, встречаются крайне редко.
Осложнения
Наиболее распространенная проблема рассматриваемого заболевания – косметические дефекты. Серьезные осложнения, представляющие угрозу для жизни больного, возникают редко. К ним относятся нарушение мозгового кровообращения при гемиатрофии лица, ишемия и гангрена пальцев рук при феномене Рейно, выраженные контрактуры суставов, инвалидизирующие пациента. Через несколько лет после дебюта болезни могут развиться тяжелые поражения внутренних органов – фиброз легких, легочная гипертензия, фиброз миокарда, перикардит, стриктуры пищевода, острая нефропатия, почечная недостаточность.
Диагностика
Пациентов с очаговой склеродермией курируют врачи ревматологи и дерматологи. При постановке диагноза учитывается клиническая картина, семейный анамнез. Все методы диагностики направлены в первую очередь на определение степени вовлечения внутренних органов и исключение системной склеродермии. С этой целью применяются следующие исследования:
- Лабораторные. В анализах крови выявляются эозинофилия, повышение уровня ревматоидного фактора, гаммаглобулинов, высокие титры антицентромерных антител и антинуклеарного фактора (АНФ). Наличие антител к топоизомеразе (анти-Scl 70) свидетельствует в пользу системного процесса. При развитии «склеродермической почки» в моче появляются белок и эритроциты.
- Инструментальные. При капилляроскопии наблюдается дилатация капилляров без участков некроза. По данным ФЭГДС могут встречаться признаки эзофагита, стриктуры пищевода. При фиброзе миокарда на ЭКГ иногда обнаруживаются нарушения ритма сердца, на ЭхоКГ – зоны гипокинеза, выпот в перикардиальную полость. На рентгенографии или компьютерной томографии легких отмечаются интерстициальные изменения.
- Гистологическое исследование биоптата кожи. Заключительный этап, позволяющий достоверно поставить диагноз. Проводится при сомнительных результатах предыдущих исследований. Характерны следующие признаки - инфильтрация лимфоцитами, плазмоцитами и эозинофилами в ретикулярном слое дермы, утолщенные коллагеновые пучки, набухание и склероз сосудистой стенки, атрофия эпидермиса, сальных и потовых желез.
Очаговую склеродермию дифференцируют с другими формами склеродермии (системной, склеродермой Бушке), дерматологическим заболеваниями (саркоидозом кожи, липонекробиозом, склеродермоподобной формы поздней кожной порфирии, базально-клеточным раком), поражением мягких тканей (панникулитом, липодерматосклерозом, эозинофильным фасциитом). В дифференциальной диагностике принимают участие онкологи, гематологи.
Лечение
Этиотропной терапии не существует. Метод лечения и вид лекарственного средства необходимо подбирать с учетом формы заболевания, тяжести течения и локализации очагов. При линейной и бляшечной формах используются топические глюкокортикостероиды высокой и сверхвысокой активности (бетаметазон, триамцинолон), синтетические аналоги витамина Д. При выраженной индурации кожи эффективны аппликации с диметилсульфоксидом. В случае поражений внутренних органов с целью уменьшения фиброзообразования назначаются пеницилламин и инъекции гиалуронидазы.
При неглубоких процессах хорошим терапевтическим действием обладает ПУВА-терапия, которая включает облучение кожи ультрафиолетовыми волнами длинного спектра с одновременным пероральным или наружным применением фотосенсибилизаторов. Тяжелое поражение кожи служит показанием к применению иммунодепрессантов (метотрексата, такролимуса, микофенолата), синдром Рейно - блокаторов кальциевых каналов (нифедипина) и препаратов, улучшающих микроциркуляцию (пентоксифиллина, ксантинола никотината). При склероатрофическом лихене проводится низкоинтенсивная лазеротерапия. В случае развития контрактур суставов, значительно затрудняющих движения, или грубых деформаций скелета и косметических дефектов лица требуется хирургическая операция.
Профилактика и прогноз
В подавляющем большинстве случаев очаговая склеродермия имеет доброкачественное течение. Правильно подобранная терапия позволяет добиться регресса симптомов. Иногда наступают спонтанные ремиссии заболевания. Неблагоприятные исходы возникают при тяжелых формах (прогрессирующей гемиатрофии лица, пансклеротической инвалидизирующей склеродермии), а также поражении внутренних органов. Эффективных методов профилактики не разработано. Рекомендуется избегать или максимально ограничить контакт кожи с химическими соединениями (кремнием, сицилием, хлорвинилом, нефтепродуктами, органическими растворителями, пестицидами, эпоксидной смолой).
2. Ревматические заболевания/ Под ред. Дж.Х. Клиппела, Дж.Х. Стоуна, Л.Дж. Кроффорд, П.Х. Уайт – 2012.
3. Диффузные болезни соединительной ткани: руководство для врачей/ под ред. проф. Мазурова В.И. –2009.
Склеродермия — поражение кожи, характеризующееся её диффузным или ограниченным уплотнением с последующим развитием фиброза и атрофии поражённых участков. К склеродермической группе болезней относят системную склеродермию, ограниченные формы склеродермии, склеродермию Бушке, эозинофильный фасциит, перекрёстные синдромы, мультифокальный фиброз, индуцированную склеродермию (в т.ч. паранеопластическую), а также псевдосклеродермические синдромы.
• Склеродермия очаговая (склеродермия ограниченная) — заболевание неясной этиологии, характеризующееся ограниченным поражением участков кожи. Висцеральные поражения не характерны •• Склеродермия бляшечная (sclerodermia placata) — участки поражения имеют форму крупных округлых плотных бляшек, окружённых полоской сиреневого цвета •• Склеродермия каплевидная (sclerodermia guttata, болезнь белых пятен, лихен атрофический первичный, лихен плоский атрофический, лихен склеротический атрофический, Цумбуша белый лихен, Чиллага болезнь) — участки поражения в виде многочисленных мелких округлых слегка запавших перламутрово-белых пятен, локализующихся на верхней части туловища •• Склеродермия кольцевидная (sclerodermia anularis) — очаги поражения в форме колец, локализованы на коже голеней, сопровождается слоновостью •• Склеродермия линейная (sclerodermia linearis) характеризуется полосовидными участками поражения, чаще в области лба
• Склеродермия отёчная (болезнь Бушке, склерема апоневротическая доброкачественная, склередема взрослых) — диффузный, быстро развивающийся болезненный плотный отёк кожи и подкожной клетчатки лица и шеи, возникающий после какого-либо инфекционного заболевания (предполагают этиологическую роль стрептококковой инфекции) и распространяющийся на верхнюю часть туловища, плечи и предплечья (поражение дистальных отделов конечностей не типично). Патоморфологически характеризуется набуханием коллагеновых волокон в глубоких слоях дермы и в подкожной клетчатке. Внутренние органы в процесс не вовлекаются. Синдром Рейно не характерен.
• Склеродермия системная (sclerodermia systemica, склеродермия генерализованная, склеродермия диффузная, склеродермия универсальная, склероз системный прогрессирующий) — системное заболевание соединительной ткани, характеризующееся воспалением и прогрессирующим фиброзом кожи и внутренних органов.
• Эозинофильный фасциит — заболевание, характеризующееся склеродермоподобным изменением кожи, возникающим на фоне эозинофилии (типичны изменения в виде «апельсиновой корки» или «желобков» по ходу запавших вен). Развиваются сгибательные контрактуры суставов. Постановке правильного диагноза способствует биопсия кожно-мышечного лоскута.
• Индуцированная склеродермия •• Обусловленная длительным контактом с кремнием, хлорвинилом •• Вызванная приёмом L-триптофана или токсичного оливкового масла, содержащего олеоанилин (эти клинические формы исчезли после устранения провоцирующих агентов) •• Спровоцированная блеомицином, пентазином •• Склеродермоподобный синдром у женщин с силиконовыми протезами молочных желёз.
• Перекрёстные синдромы системной склеродермии с другими системными заболеваниями соединительной ткани (дерматомиозитом, полимиозитом, ревматоидным артритом, СКВ).
• Мультифокальный фиброз. Термин формально объединяет ряд редко встречающихся состояний, характеризующихся регионарным фиброзом •• Висцеральные, или большие, фиброзы: ретроперитонеальный, или болезнь Ормонда (фиброз «замуровывает» мочеточники, матку, реже — надпочечники, поджелудочную железу, кишечник), эндокардиальный, медиастинальный •• Поверхностные, или малые, фиброзы: контрактура Дюпюитрена, зоб Риделя, псевдоопухоль орбит глаз, болезнь Пейрони, келоид.
• Паранеопластические склеродермоподобные синдромы (см. Синдром паранеопластический).
• Псевдосклеродермическая симптоматика (уплотнения кожи или заострение черт лица) сопровождает ряд метаболических нарушений и эндокринных заболеваний. Для постановки правильного диагноза необходим критический анализ синдромов, сопутствующих кожным изменениям •• Метаболические нарушения: поздняя кожная порфирия, фенилкетонурия, болезнь Вильсона–Коновалова, системный амилоидоз •• Эндокринопатии: синдром Вернера–Моррисона, синдром Симмондса–Глински, синдром Ротмунда, гипотиреоз.
МКБ-10 • M34 Системный склероз • L94 Другие локализованные изменения соединительной ткани.
Код вставки на сайт
Склеродермия — поражение кожи, характеризующееся её диффузным или ограниченным уплотнением с последующим развитием фиброза и атрофии поражённых участков. К склеродермической группе болезней относят системную склеродермию, ограниченные формы склеродермии, склеродермию Бушке, эозинофильный фасциит, перекрёстные синдромы, мультифокальный фиброз, индуцированную склеродермию (в т.ч. паранеопластическую), а также псевдосклеродермические синдромы.
• Склеродермия очаговая (склеродермия ограниченная) — заболевание неясной этиологии, характеризующееся ограниченным поражением участков кожи. Висцеральные поражения не характерны •• Склеродермия бляшечная (sclerodermia placata) — участки поражения имеют форму крупных округлых плотных бляшек, окружённых полоской сиреневого цвета •• Склеродермия каплевидная (sclerodermia guttata, болезнь белых пятен, лихен атрофический первичный, лихен плоский атрофический, лихен склеротический атрофический, Цумбуша белый лихен, Чиллага болезнь) — участки поражения в виде многочисленных мелких округлых слегка запавших перламутрово-белых пятен, локализующихся на верхней части туловища •• Склеродермия кольцевидная (sclerodermia anularis) — очаги поражения в форме колец, локализованы на коже голеней, сопровождается слоновостью •• Склеродермия линейная (sclerodermia linearis) характеризуется полосовидными участками поражения, чаще в области лба
• Склеродермия отёчная (болезнь Бушке, склерема апоневротическая доброкачественная, склередема взрослых) — диффузный, быстро развивающийся болезненный плотный отёк кожи и подкожной клетчатки лица и шеи, возникающий после какого-либо инфекционного заболевания (предполагают этиологическую роль стрептококковой инфекции) и распространяющийся на верхнюю часть туловища, плечи и предплечья (поражение дистальных отделов конечностей не типично). Патоморфологически характеризуется набуханием коллагеновых волокон в глубоких слоях дермы и в подкожной клетчатке. Внутренние органы в процесс не вовлекаются. Синдром Рейно не характерен.
• Склеродермия системная (sclerodermia systemica, склеродермия генерализованная, склеродермия диффузная, склеродермия универсальная, склероз системный прогрессирующий) — системное заболевание соединительной ткани, характеризующееся воспалением и прогрессирующим фиброзом кожи и внутренних органов.
• Эозинофильный фасциит — заболевание, характеризующееся склеродермоподобным изменением кожи, возникающим на фоне эозинофилии (типичны изменения в виде «апельсиновой корки» или «желобков» по ходу запавших вен). Развиваются сгибательные контрактуры суставов. Постановке правильного диагноза способствует биопсия кожно-мышечного лоскута.
• Индуцированная склеродермия •• Обусловленная длительным контактом с кремнием, хлорвинилом •• Вызванная приёмом L-триптофана или токсичного оливкового масла, содержащего олеоанилин (эти клинические формы исчезли после устранения провоцирующих агентов) •• Спровоцированная блеомицином, пентазином •• Склеродермоподобный синдром у женщин с силиконовыми протезами молочных желёз.
• Перекрёстные синдромы системной склеродермии с другими системными заболеваниями соединительной ткани (дерматомиозитом, полимиозитом, ревматоидным артритом, СКВ).
• Мультифокальный фиброз. Термин формально объединяет ряд редко встречающихся состояний, характеризующихся регионарным фиброзом •• Висцеральные, или большие, фиброзы: ретроперитонеальный, или болезнь Ормонда (фиброз «замуровывает» мочеточники, матку, реже — надпочечники, поджелудочную железу, кишечник), эндокардиальный, медиастинальный •• Поверхностные, или малые, фиброзы: контрактура Дюпюитрена, зоб Риделя, псевдоопухоль орбит глаз, болезнь Пейрони, келоид.
• Паранеопластические склеродермоподобные синдромы (см. Синдром паранеопластический).
• Псевдосклеродермическая симптоматика (уплотнения кожи или заострение черт лица) сопровождает ряд метаболических нарушений и эндокринных заболеваний. Для постановки правильного диагноза необходим критический анализ синдромов, сопутствующих кожным изменениям •• Метаболические нарушения: поздняя кожная порфирия, фенилкетонурия, болезнь Вильсона–Коновалова, системный амилоидоз •• Эндокринопатии: синдром Вернера–Моррисона, синдром Симмондса–Глински, синдром Ротмунда, гипотиреоз.
МКБ-10 • M34 Системный склероз • L94 Другие локализованные изменения соединительной ткани.
Авторы: Ахмина Н.И. 1 , Уткин П.С. 2 , Шалатонин М.П. 3 , Чабаидзе Ж.Л. 1 , Дементьев А.А. 1 , Заплатников А.Л. 1
1 ФГБОУ ДПО РМАНПО Минздрава России, Москва, Россия
2 ГБУЗ ДИКБ No 6 ДЗМ, Москва, Россия
3 ГБУЗ «Морозовская ДГКБ ДЗМ», Москва, Россия
Склеродермия — редкая патология в педиатрической практике, хотя и занимает 3-е место после ювенильного идиопатического артрита и системной красной волчанки среди всех ревматических заболеваний детского возраста. При этом варианты врожденной склеродермии (ВС) рассматриваются как казуистика, так как в мировой литературе до настоящего времени имеются лишь единичные описания таких случаев. Этиология ВС неизвестна. Основными патогенетическими звеньями заболевания являются: повреждение сосудистого русла, дисфункция иммунной системы с нарушением регуляции высвобождения цитокинов и измененный синтез коллагена с пролиферацией фибробластов и последующим развитием фиброза. В статье приведено описание клинического случая склеродермии у новорожденного ребенка с врожденной локализованной нодулярной формой, клинические проявления которой были выявлены уже при рождении. Описаны трудности верификации диагноза на основании клинических и рутинных лабораторно-инструментальных исследований. Подчеркивается роль современных морфологических исследований, ввиду отсутствия специфических лабораторных тестов, в установлении диагноза склеродермии у новорожденного ребенка. Также представлен краткий обзор литературы, посвященной проблеме ВС.
Ключевые слова: врожденная склеродермия, дети, диагностика, дифференциальная диагностика, морфологические исследования.
N.I. Akhmina 1 , P.S. Utkin 2 , M.P. Shalatonin 3 , Zh.L. Chabaidze 1 , A.A. Dement’ev 1 , A.L. Zaplatnikov 1
1 Russian Medical Academy of Continuous Professional Education, Moscow,
2 Children’s Infectious Clinical Hospital No. 6, Moscow, Russian Federation
3 Morozov Children’s City Clinical Hospital, Moscow, Russian Federation
Scleroderma is a rare condition in pediatrics although it ranks third (after juvenile idiopathic arthritis and systemic lupus erythematosus) among rheumatic diseases in children. Congenital scleroderma is considered casuistic since only single case reports are available in publications worldwide. The etiology of congenital scleroderma remains elusive. The principal pathogenic mechanisms are vascular network impairment, immune system dysfunction with abnormal regulation of cytokine release and altered collagen synthesis with fibroblast proliferation and fibrosis. This paper describes a newborn girl with congenital localized nodular scleroderma whose clinical presentations were detected at birth. The authors discuss difficulties with the diagnosis based on clinical and routine lab and instrumental tests. The role of modern morphological studies (given the lack of specific lab tests) to diagnose scleroderma in a newborn is emphasized. Finally, this paper provides a brief review of published data on congenital scleroderma.
Keywords: congenital scleroderma, children, diagnostics, differential diagnosis, morphological studies.
Введение
Ювенильной склеродермией (ЮС) называют склеродермию, при которой клинические проявления манифестируют в возрасте до 18 лет. ЮС является редкой ревматологической патологией детского возраста. В то же время среди всех аутоиммунных заболеваний соединительной ткани у детей ЮС занимает 3-е место после ювенильного идиопатического артрита и системной красной волчанки. Этиология ЮC остается неизвестной. Основными патогенетическими звеньями заболевания являются: повреждение сосудистого русла, дисфункция иммунной системы с нарушением регуляции высвобождения цитокинов и измененный синтез коллагена с пролиферацией фибробластов и последующим развитием фиброза. Специфические лабораторные тесты, на основании которых можно было бы верифицировать заболевание, до настоящего времени не разработаны. Диагноз устанавливается, как правило, на основании клинико-анамнестических данных и с учетом характерных гистологических признаков, если были проведены морфологические исследования. Учитывая клинические особенности, выделяют системную и ограниченную ЮС. Системная ЮС характеризуется развитием склероза кожи и поражением внутренних органов. При ограниченной ЮС имеет место преимущественное поражение кожи и/или подлежащих тканей с развитием в дальнейшем контрактур и значительно реже — с повреждением костных структур [1–5]. Особо следует отметить, что в мировой литературе [6–11] имеются описания единичных случаев склеродермии, клинические проявления которой были обнаружены уже при рождении детей (врожденная склеродермия, ВС). Казуистическая частота встречаемости ВС и трудности ее диагностики явились основанием для настоящей публикации.
Клиническое наблюдение
Девочка М. родилась от 39-летней женщины, от 5-й беременности, от 3-х оперативных родов на 39-й неделе гестации. Предыдущие беременности: 1-я — срочные роды; 2-я и 3-я — медицинские аборты; 4-я — оперативные своевременные роды; 5-я — настоящая беременность беременность, которая протекала с токсикозом в I триместре, острой респираторной инфекцией без подъема температуры во II триместре и преэклампсией 1-й степени в III триместре.
В семейном анамнезе: у матери имеются желчнокаменная болезнь, миопия слабой степени, варикозная болезнь вен нижних конечностей, эрозия шейки матки. Во время беременности мать в плановом порядке обследована на сифилис, гепатиты В и С, ВИЧ-инфекцию: результаты отрицательные. Отцу 40 лет, курит. Заболевания кожи и онкологическая патология в семейном анамнезе не установлены. Аллергический анамнез не отягощен.
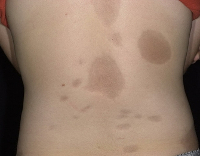
При рождении: масса 3100 г, длина 50 см, окружность головы 35 см, окружность груди 32 см. Оценка по шкале Апгар 7–8 баллов. Состояние ребенка при рождении средней степени тяжести. При рождении на туловище (грудь, живот, спина), на конечностях и на лице отмечены узловатые образования от 3–5 мм до 25–35 мм в диаметре, плотные, возвышающиеся над уровнем кожи, местами сливающиеся, кожа над ними гиперемирована с синюшным оттенком (рис. 1). Дыхательные и гемодинамические нарушения отсутствовали. В неврологическом статусе — синдром угнетения с элементами возбуждения.

На 2-е сутки жизни девочка переведена из роддома в ГБУЗ ДИКБ № 6 ДЗМ. При поступлении, помимо описанных элементов на коже, был отмечен также гепатолиенальный синдром: печень (по Курлову) +1,5 см, +3,0 см, +1 см; селезенка +2 см. Рефлексы новорожденного вызывались в полном объеме. Учитывая клинические проявления, намеченный план обследования был направлен на проведение дифференциального диагноза между внутриутробными инфекциями, врожденным лейкозом и различными формами генодерматозов. Проведенное комплексное обследование позволило исключить врожденные инфекции и гемобластозы.
На фоне проводимой терапии (амоксициллин/клавуланат, внутривенное введение глюкозосолевых растворов, инозин, пиридоксин, никотинамид, рибофлавин) была отмечена некоторая положительная динамика. Кожные проявления стали менее выраженными, некоторые узлы в области спины, груди, конечностей купировались, на их месте остались пигментные пятна. Крупные узлы имели тенденцию к уменьшению в размерах. В динамике клинических проявлений не было отмечено инфекционного процесса, лабораторные показатели оставались в пределах референсных значений. Аппетит у девочки был хороший, питание усваивала в полном объеме, не срыгивала, весовая кривая — положительная.
Однако диагноз оставался неясным, несмотря на привлечение к курации ребенка врачей разных специальностей. Учитывая это, была проведена биопсия узла с последующим морфологическим исследованием, которое позволило выявить типичные признаки склеродермии (рис. 2). Принимая во внимание сроки манифестации заболевания и клинико-морфологические особенности, верифицирована ограниченная нодулярная ВС.

Вопросы, связанные с истинной частотой встречаемости ВС, активно обсуждаются в течение последних 10–15 лет. Это обусловлено тем, что клинические симптомы ВС, обнаруженные при рождении, нередко в течение длительного времени ошибочно трактуются как проявления других заболеваний. Неточная диагностика приводит к существенной задержке назначения адекватной терапии и, как следствие, к прогрессированию заболевания и высокому риску развития осложнений [6–11]. Так, по данным M. Mansour et al. [6], которые проводили международное мультицентровое исследование на протяжении 16 лет (2001–2017 гг.) и смогли выявить 25 педиатрических пациентов с ВС, среднее время от рождения детей с клиническими проявлениями ВС до постановки диагноза составило 2,9 года. При этом у 12 из 25 пациентов к этому времени уже имели место внекожные проявления склеродермии (скелетно-мышечные повреждения отмечены у 80% пациентов, неврологические — у 62%). Аналогичные результаты задержки постановки диагноза представлены и при описании единичных наблюдений ВС [7–11]. Учитывая, что в основе склеродермии лежат аутоиммунные механизмы, а также то, что по-прежнему не исключена вероятность стадийности повреждений различных органов и систем при склеродермии (на ранних стадиях — кожа и подлежащие ткани (ограниченная склеродермия), на поздних — внутренние органы (системная склеродермия)), очень важно своевременно установить диагноз и назначить адекватную терапию [1, 12–16].
Заключение
Все вышесказанное определяет необходимость включать ВС в перечень вероятных заболеваний в тех случаях, когда у новорожденного на коже туловища, и/или конечностей, и/или головы имеют место различные по величине округлые участки гиперемии с цианотичным оттенком на узловатых образованиях, возвышающихся над уровнем кожи. Учитывая, что специфические лабораторные тесты на ВС до настоящего времени не разработаны, окончательная верификация заболевания основывается на характерных гистологических признаках при морфологическом исследовании микропрепаратов, полученных после проведения биопсии измененных участков кожи и подлежащих тканей.
Сведения об авторах:
Ахмина Наталия Ивановна — д.м.н., профессор кафедры неонатологии им. проф. В.В. Гаврюшова ФГБОУ ДПО РМАНПО Минздрава России; 123995, Россия, г. Москва, ул. Баррикадная, д. 2/1; ORCID iD 0000-0002-5870-4938.
Уткин Петр Сергеевич — врач-дерматолог ГБУЗ ДИКБ № 6 ДЗМ; 125438, Россия, г. Москва, 3-й Лихачевский пер., д. 2Б; ORCID iD 0000-0001-5934-8480.
Чабаидзе Жужуна Лазаревна — к.м.н., доцент кафедры неонатологии им. проф. В.В. Гаврюшова ФГБОУ ДПО РМАНПО Минздрава России; 123995, Россия, г. Москва, ул. Баррикадная, д. 2/1; ORCID iD 0000-0002-2192-796X.
Дементьев Александр Анатольевич — к.м.н., доцент кафедры неонатологии им. проф. В.В. Гаврюшова ФГБОУ ДПО РМАНПО Минздрава России; 123995, Россия, г. Москва, ул. Баррикадная, д. 2/1; ORCID iD 0000-0002-7640-1172.
Заплатников Андрей Леонидович — д.м.н., профессор, проректор по учебной работе, заведующий кафедрой неонатологии им. проф. В.В. Гаврюшова, профессор кафедры педиатрии им. акад. Г.Н. Сперанского ФГБОУ ДПО РМАНПО Минздрава России; 123995, Россия, г. Москва, ул. Баррикадная, д. 2/1; ORCID iD 0000-0003-1303-8318.
Прозрачность финансовой деятельности: никто из авторов не имеет финансовой заинтересованности в представленных материалах или методах.
Конфликт интересов отсутствует.
Статья поступила 09.03.2022.
Поступила после рецензирования 01.04.2022.
Принята в печать 26.04.2022.
About the authors:
Nataliya I. Akhmina — Dr. Sc. (Med.), professor of the Prof. V.V. Gavryushov Department of Neonatology, Russian Medical Academy of Continuous Professional Education; 2/1, Barrikadnaya str., Moscow, 125993, Russian Federation; ORCID iD 0000-0002-5870-4938.
Petr S. Utkin — dermatologist, Children’s Infectious Clinical Hospital No. 6; 2B, 3rd Likhachevskiy lane, Moscow, 125438, Russian Federation; ORCID iD 0000-0001-5934-8480.
Zhuzhuna L. Chabaidze — C. Sc. (Med.), associate professor of the Prof. V.V. Gavryushov Department of Neonatology, Russian Medical Academy of Continuous Professional Education; 2/1, Barrikadnaya str., Moscow, 125993, Russian Federation; ORCID iD 0000-0002-2192-796X.
Aleksandr A. Dement’ev — C. Sc. (Med.), associate professor of the Prof. V.V. Gavryushov Department of Neonatology, Russian Medical Academy of Continuous Professional Education; 2/1, Barrikadnaya str., Moscow, 125993, Russian Federation; ORCID iD 0000-0002-7640-1172.
Andrey L. Zaplatnikov — Dr. Sc. (Med.), Professor, Vice-chancellor for Instructional Work, Head of the Prof. N.N. Gavryushov Department of Neonatology, professor of the Acad. G.N. Speranskiy Department of Pediatrics, Russian Medical Academy of Continuous Professional Education; 2/1 Barrikadnaya str., Moscow, 125993, Russian Federation; ORCID iD 0000-0003-1303-8318.
There is no conflict of interests.
Контент доступен под лицензией Creative Commons «Attribution» («Атрибуция») 4.0 Всемирная.
Сотрудники Санкт-Петербургского государственного педиатрического медицинского университета подготовили статью посвященную вопросам терапии ювенильной локализованной склеродермии. Тактика лечения определяется площадью и глубиной очагов кожного поражения, появлением и распространением новых очагов, наличием внекожных признаков болезни. Основой терапии ЮЛС служат топические и системные иммуносупрессанты. Антибиотикотерапия не показана. Для оценки эффективности терапии рекомендовано использовать клинические шкалы (LoSCAT), ультразвуковое исследование, инфракрасную термографию и магнитно-резонансную томографию.
Заболеваемость локализованной склеродермией в Российской Федерации, по последним данным, составляет 3,3–3,7 на 100 тыс. населения, у детей в возрасте 15–17 лет — 4,3–5,9 на 100 тыс. населения. Детскими ревматологами принято использовать классификацию ювенильной локализованной склеродермии (ЮЛС) от 2006 г., в соответствии с которой выделяют очаговую, линейную, генерализованную, пансклеротическую и смешанную склеродермию. Отличительной особенностью ЮЛС является высокая частота внекожных проявлений (изменения со стороны опорно-двигательного аппарата, глаз, нервной системы) — у 15–20% больных, что требует междисциплинарного подхода к их ведению.
Патогенез заболевания связан с иммунными нарушениями, приводящими к активации профибротических медиаторов и чрезмерной выработке коллагена. Учитывая значимую роль иммунной системы в развитии заболевания, большинство препаратов нацелено на подавление иммунной активности. При этом основной целью лечения является достижение статуса неактивной болезни, а в последующем — клинической ремиссии (12 мес неактивной болезни). Выбор тактики лечения зависит от формы локализованной склеродермии и степени активности заболевания. Общепризнанно, что широко распространяющиеся, прогрессирующие, глубокие очаги, очаги, пересекающие суставы, находящиеся на косметически чувствительных зонах, должны лечиться системными иммуносупрессивными препаратами. Для лечения очаговой склеродермии (бляшечная, каплевидная морфея) на ранних этапах болезни можно применять (монотерапия, комбинации) топические препараты, в числе которых находятся кальципотриол, местные глюкокортикостероиды, имиквимод и ингибиторы кальциневрина.
Топическая терапия
Топические глюкокортикостероиды. Несмотря на широкое применение топических глюкокортикостероидов в клинической практике, доказательства эффективности препаратов этой группы при локализованной склеродермии отсутствуют. Местные глюкокортикостероиды подавляют воспаление и ингибируют пролиферацию фибробластов, оказывая антифибротическое воздействие. Препараты высокой (мометазона фуроат, метилпреднизолона ацепонат, бетаметазона дипропионат) или очень высокой (клобетазола пропионат) степени активности могут применяться при очаговой склеродермии на туловище, умеренной степени активности (алклометазон, триамцинолона ацетонид) — при поражении лица. Эффективность местных глюкокортикостероидов при формах склеродермии, где показана системная терапия, включая линейную склеродермию, не исследована. Ввиду потенциальных рисков развития атрофии кожи при длительном применении глюкокортикостероидов их ежедневное использование должно быть ограничено.
Топические ингибиторы кальциневрина. Ингибиторы кальциневрина (пимекролимус крем 1%, такролимус мазь 0,03 и 0,1%) обладают иммуносупрессивным и противовоспалительным эффектом. Механизм действия обусловлен блокадой кальциневрина и подавлением T-хелперной активности, что приводит к снижению интенсивности выработки провоспалительных цитокинов.
В двойном слепом плацебоконтролируемом исследовании топического такролимуса 0,1% у 10 пациентов с очаговой склеродермией (средний возраст 44 года, длительность заболевания 1–9 лет) была доказана эффективность препарата. После 12 нед лечения в основной группе отмечено разрешение ранних склеротических очагов и смягчение длительно существующих очагов, тогда как в группе плацебо очаги склеродермии не претерпевали изменений. В двух других плацебоконтролируемых открытых исследованиях также была отмечена эффективность топического такролимуса 0,1% при очаговой склеродермии. В первом исследовании оценивали гистологическую картину, обнаружено снижение лимфоцитарной инфильтрации очага склеродермии через 4 мес после начала терапии. Во втором исследовании дополнительно применяли окклюзионные повязки, отмечено исчезновение ранних воспалительных кожных изменений и смягчение, снижение интенсивности поздних склеротических изменений.
Топический имиквимод. Имиквимод — агонист toll-like рецептора 7 (TLR7). Имиквимод связывается с TLR7 на поверхности антигенпрезентирующих клеток и активирует его, запуская каскад иммунных реакций, влияющих на врожденный и приобретенный иммунитет.
Исследование 5% крема имиквимод, который применяли 3–7 раз/нед у 12 пациентов (возраст 6–77 лет) с различными формами локализованной склеродермии в течение 6 мес, показало клиническое улучшение в виде снижения интенсивности эритемы и диспигментации, уменьшения плотности очагов с гистологическим подтверждением — уменьшением толщины дермы. Использование имиквимода у детей с ограниченной склеродермией в течение 9 мес сопровождалось уменьшением степени выраженности эритемы, диспигментации и уменьшением утолщения кожи.
Фототерапия. Лечение светом ультрафиолетового спектра представляет собой альтернативу системной терапии при глубокой или линейной склеродермии для пациентов старше 12 лет. Действие фототерапии опосредовано снижением продукции коллагена, выработки провоспалительных цитокинов (IL 6, IL 8, TNF ) и подавлением апоптоза эндотелиальных клеток. Результат применения фототерапии у взрослых пациентов позволяет предполагать, что этот способ лечения может быть применен и у детей, однако безопасность метода, оптимальные дозы и схемы лечения остаются неизвестными.
Согласно рекомендациям T. Constantin и соавт., детям старше 12 лет с маленькими поверхностными непрогрессирующими очагами склеродермии, не пересекающими область сустава и располагающимися в косметически нечувствительных зонах, возможно проведение фототерапии, предпочтительно UVA-1 или UVB (ультрафиолетовое излучение типа В — электромагнитное излучение с диапазоном волны 280–315 нм). PUVA-терапия (методика фототерапии, основанная на сочетанном применении фотоактивного вещества псорален (Р), принимаемого внутрь, и последующего облучения длинноволновыми ультрафиолетовыми лучами, UVA) детям противопоказана.
Системная иммуносупрессивная терапия. Терапия топическими препаратами и фототерапия без системных иммуносупрессантов целесообразна только при поверхностных небольших очагах склеродермии, которые не прогрессируют в числе и размерах и не пересекают область сустава, а также находятся в косметически нечувствительных зонах. В любых других случаях показана системная иммунносупрессивная терапия.
Метотрексат. Согласно результатам наблюдательных и клинических исследований, а также консенсусу, метотрексат является первой линией терапии линейной, генерализованной и пансклеротической склеродермии. Согласно консенсусу детских ревматологов 2019 г., всем пациентам с выраженными косметическими и/или функциональными нарушениями может быть рекомендована терапия метотрексатом из расчета 15 мг/м2 подкожно или перорально. В 2/3 случаев терапия метотрексатом позволяет достичь неактивной фазы болезни. Результаты длительного (в среднем 30 лет) наблюдения за пациентами с локализованной склеродермией демонстрируют, что при ювенильном дебюте болезни признаки активного заболевания во взрослом возрасте имеют около 90% пациентов, необратимые осложнения — более половины. В связи с этим ожидается, что наилучшие результаты лечения метотрексатом могут быть достигнуты при своевременном начале лечения — до появления признаков кожного повреждения (необратимых изменений). Важно также подчеркнуть, что длительная ремиссия после приема метотрексата чаще отмечается у пациентов, получавших лечение в течение более чем 12 мес после достижения клинической ремиссии. Таким образом, как только достигается приемлемое клиническое улучшение, назначение метотрексата должно быть продолжено в течение как минимум 12 мес, хотя часто используются и более длительные схемы лечения. В течение первых 3 мес лечения в качестве дополнения к терапии метотрексатом следует использовать курс глюкокортикостероидов. Эффективность и безопасность системных глюкокортикостероидов в активной фазе ЮЛС подтверждены в ряде исследований.
Терапия при неэффективности метотрексата. Согласно результатам британского национального аудита по оценке и лечению локализованной склеродермии, 95,5% из 149 пациентов получали метотрексат как первую линию терапии. Каждый третий пациент (34%) получал два болезньмодифицирующих лекарственных препарата или более, т. е. не достиг ремиссии на фоне лечения метотрексатом, что потребовало усиления терапии. При неэффективности метотрексата, при рецидивировании болезни после клинической ремиссии (прогрессирование кожных очагов, тяжелые внекожные проявления) или в случае непереносимости метотрексата может быть использован микофенолата мофетил (MMФ) в дозе 500–1000 мг/м2 как в режиме монотерапии, так и в комбинации с метотрексатом. MMФ является наиболее часто используемым препаратом второй линии терапии (у 89,5% больных) и обладает такой же эффективностью в отношении кожного статуса, как и монотерапия метотрексатом, но оказывает меньшее влияние на артрит по сравнению с метотрексатом.
Тоцилизумаб — моноклональное антитело к рецепторам IL 6. Препарат показан для лечения полиартикулярного и системного ювенильного идиопатического артрита. Ингибирование IL 6 может быть эффективной мишенью терапии больных локализованной склеродермией, что обусловлено повышенной концентрацией этого цитокина в сыворотке крови и корреляцией с числом линейных поражений. В фазе II клинических испытаний тоцилизумаб показал хорошую эффективность в отношении кожного и легочного поражения у взрослых пациентов с системной склеродермией.
Абатацепт — растворимый рекомбинантный белок, который специфически связывается с молекулами CD80 и CD86 на поверхности антигенпрезентирующих клеток и блокирует таким образом их взаимодействие с рецепторами CD28 Т-лимфоцитов. Абатацепт разрешен для лечения детей с полиартикулярным ювенильным идиопатическим артритом [51]. Вместе с тем абатацепт является перспективным препаратом для лечения поражения кожи при системной склеродермии; проводятся клинические испытания у взрослых пациентов. В связи с тем, что патогенетические механизмы кожного поражения при системной и локализованной склеродермии схожи, абатацепт может быть использован при локализованной склеродермии. Опубликованы данные успешного лечения абатацептом взрослых пациентов с локализованной склеродермией.
Физиотерапия. Ортопедические осложнения, такие как укорочение, атрофия конечности, контрактуры суставов, возникают примерно у 30–50% пациентов с линейной склеродермией конечности. Физиотерапия, лечебная физкультура в сочетании с системной иммуносупрессивной терапией рекомендованы для детей с линейной, глубокой склеродермией и всем пациентам со склеродермией при снижении объема движений в суставах и снижении мышечной силы, чтобы избежать и/или минимизировать возникновение контрактур суставов. При поражении кистей и пальцев эффективным методом профилактики являются ежедневные домашние упражнения с пассивным разгибанием и растяжкой.
Хирургические методы лечения. Оперативные вмешательства при любых формах склеродермии не рекомендуется выполнять в активную фазу болезни. Хирургическая коррекция возможна при контрактурах суставов и деформациях, приводящих к нарушению двигательной активности и качества жизни. Эффективность метода в настоящее время изучена недостаточно. Основной проблемой является возможное возобновление активности и прогрессирование склеродермии после оперативного вмешательства.
Косметические проблемы, связанные со склеродермией в области лица, оказывают значительное влияние на психологическое состояние пациентов и качество их жизни. Методом решения этих проблем являются пластические операции. На сегодняшний день чаще всего применяют аллогенную трансплантацию жировой ткани. В ретроспективном исследовании 10 пациентов с ювенильной склеродермией области лица была выполнена пластическая коррекция, после которой все они отметили, что рекомендовали бы другим пациентам данный метод лечения.
Заключение. Детям с ЮЛС требуется своевременное и адекватное лечение для предупреждения необратимых изменений и лучшего исхода болезни. Терапия зависит от формы и активности склеродермии. При очаговой склеродермии широко и успешно применяют топические препараты. Системная иммуносупрессивная терапия может потребоваться при обширных очагах, глубоком поражении, поражении косметически чувствительных зон, быстром прогрессировании старых очагов и появлении новых, внекожных, проявлений. Метотрексат — препарат первой линии терапии при линейной, генерализованной, пансклеротической, смешанной форме. Учитывая, что терапевтический эффект препарата может быть достигнут в течение 2–3 мес, для быстрого подавления воспалительной активности в сочетании с метотрексатом короткими курсами могут быть применены глюкокортикостероиды. При достижении ремиссии заболевания рекомендуется продолжить терапию метотрексатом в течение не менее 12 мес. Треть пациентов не отвечают на терапию метотрексатом и требуют назначения других болезньмодифицирующих или биологических препаратов.
Читайте также: