
Лекарства при буллезном эпидермолиз
Обновлено: 25.04.2024
Фонд «Дети-Бабочки» совместно с Национальным медицинским исследовательским центром акушерства, гинекологии и перинатологии имени академика В.И. Кулакова запустили совместный проект по оказанию комплексной специализированной помощи детям с буллезным эпидермолизом и их семьям
На сегодняшний день буллезный эпидермолиз (БЭ) – неизлечимое заболевание с хроническим течением. Возможности современной медицины, по крайней мере пока, сводятся к симптоматической терапии, правильному уходу и поиску новых протоколов лечения, которые позволили бы лучше справляться с проявлениями БЭ, предотвращать (или хотя бы отодвинуть во времени) тяжелые осложнения и повысить качество жизни пациентов.
Уже почти 11 лет фонд «Дети-Бабочки» оказывает комплексную помощь пациентам с буллезным эпидермолизом и их семьям: обеспечивает медицинскую, психологическую и юридическую поддержку, медикаменты, патронаж, обучение врачей. Совместно с ФГБУ «Национальный Медицинский Исследовательский Центр акушерства, гинекологии и перинатологии имени академика В.И. Кулакова» Министерства здравоохранения Российской Федерации (ФГБУ «НМИЦ АГП им. В.И. Кулакова» МЗ РФ) фондом был инициирован специальный проект, который позволит детям с БЭ по всей России получить высококачественную медицинскую помощь.
Центр имеет большие диагностические мощности, активно использует инновационные технологии лечения, в частности, клеточную терапию. Фонд «Дети-Бабочки», в свою очередь, обладает организационными компетенциями и наработками в области ухода за детьми с буллезным эпидермолизом. Объединение усилий дает группе врачей и исследователей шанс на разработку новых протоколов лечения.
«Мы можем «подхватить» маленького пациента в самом начале его жизни и дать задел на будущее, ресурс для организма, чтобы как можно дольше избегать осложнений, таких как длительно незаживающие раны, срастание пальцев, контрактуры и тому подобное» – подчеркивает Юлия Юрьевна Коталевская, врач-генетик фонда.
В проекте участвуют дети с самой тяжелой дистрофической формой БЭ. Первый этап программы – проведение телемедицинского консилиума регионального врача и специалистов ФГБУ «НМИЦ АГП им. В.И. Кулакова» МЗ РФ. В процессе телеконсультации оценивается состояние новорожденного, согласовывается тактика медицинской помощи, определяется тактика ухода за кожей, обсуждается необходимость перевода ребенка в Москву. После дифференциальной диагностики ребенок госпитализируется в ФГБУ «НМИЦ АГП им. В.И. Кулакова» МЗ РФ, где разрабатывается индивидуальный план лечения.
Фонд финансирует собственно научную составляющую проекта, затраты на генетическую диагностику и медикаменты на весь период госпитализации. Пребывание в стационаре ребенка с мамой, необходимые исследования, лечение других заболеваний и уход производятся за счет средств ОМС. Транспортные расходы покрываются за счет региональных департаментов здравоохранения и средств благотворительного фонда «Дети-бабочки».
Программа пребывания в стационаре объединяет в себе две главные составляющие:
- правильный уход с первых дней жизни, который поможет изначально избежать серьезных (и, к сожалению, частых) ошибок в ведении пациентов с БЭ;
- терапию с применением клеточных технологий, направленную на стимуляцию процессов регенерации и поддержание стабильности кожного покрова.
Специалисты фонда курируют лечение на всех его стадиях и находятся в постоянном контакте с врачами в отделении и мамой пациента.
После поступления ребенку назначается комплексное обследование и консультация узкопрофильных специалистов. К моменту перевода пациента в отделение фонд организует доставку специальных перевязочных материалов. Дополнительно под контролем медиков маму обучают основам ухода за ребенком.
Изначально проект был рассчитан на три года, но пандемия «скорректировала» его продолжительность. Тем не менее, в 2022 году планируется публикация промежуточных данных, которые в первом приближении позволят сделать выводы об эффективности протоколов клеточной терапии.
Исследования по поиску эффективных способов борьбы с патологией кожи проводятся во всем мире. Совместный проект Фонда и ФГБУ «НМИЦ АГП им. В.И. Кулакова» МЗ РФ по созданию мультидисциплинарного подхода в сочетании с клеточными технологиями – один из шагов в этом направлении.
Что такое эксфолиативный кератолиз? Причины возникновения, диагностику и методы лечения разберем в статье доктора Похлебкиной Алевтины Алексеевны, педиатра со стажем в 6 лет.
Над статьей доктора Похлебкиной Алевтины Алексеевны работали литературный редактор Вера Васина , научный редактор Владимир Горский и шеф-редактор Маргарита Тихонова
Определение болезни. Причины заболевания
Эксфолиативный кератолиз — это очаговое симметричное шелушение кожи на ладонях, поверхности пальцев и, реже, на подошвах. Заболевание характеризуется сухостью кожи и поверхностными пузырями, заполненных воздухом.
Также эксфолиативный кератолиз называют рецидивирующей фокальной ладонной десквамацией, сухим пластинчатым дисгидрозом и рецидивирующей ладонной десквамацией.
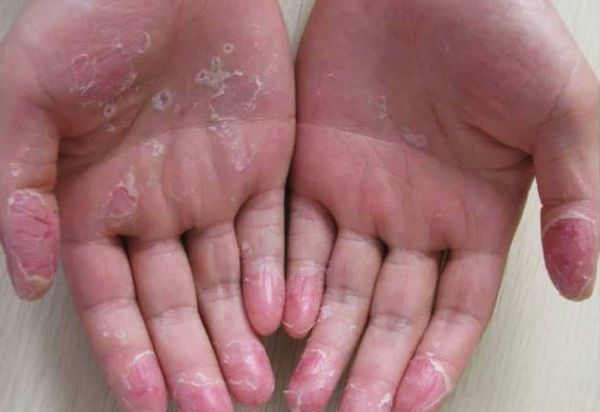
Заболевание широко распространено и часто носит хронический, но доброкачественный характер. Эксфолиативный кератолиз чаще встречается у детей и подростков, реже у взрослых. Нередко его ошибочно принимают за псориаз, экзему или хронический контактный дерматит. У людей с повышенной потливостью рук состояние ухудшается в тёплую погоду и может быть связано с гипергидрозом — усиленным потоотделением [1] .
Ранее эксфолиативный кератолиз называли дисгидротической экземой , и считалось, что заболевание вызвано нарушением работы потовых желёз. Эта связь уже опровергнута, но термин "дисгидротическая экзема" всё ещё используется [2] .
Дисгидротическая экзема, также называемая помфоликсом, может предшествовать эксфолиативному кератолизу. При этом состоянии на пальцах рук, ног, ладонях и подошвах образуются волдыри, наполненные жидкостью, и возникает сильный зуд [2] . Причина дисгидротической экземы неизвестна, но, вероятно, на развитие заболевания влияет множество факторов. В большинстве случаев причину и предрасполагающий фактор выделить невозможно [17] .
Причины эксфолиативного кератолиза
Предполагалось, что экфолиативный кератолиз может быть вызван грибковым поражением, но в дальнейших исследованиях эта гипотеза не подтвердилась.
Возможные провоцирующие факторы эксфолиативного кератолиза:
- трение и контакт с водой[3][6];
- мыло, моющие средства и растворители: химические вещества, содержащиеся в них, могут привести к появлению трещин и пузырей на руках;
- аллергия: продукты питания, загрязнение воздуха и другие вещества могут спровоцировать появление аллергии на коже;
- стресс: может не только вызвать, но и усугубить течение болезни;
- жаркий климат: при тёплой погоде потоотделение усиливается, что может привести к шелушению кожи;
- воздействие солёной воды;
- сухость кожи.
Эксфолиативный кератолиз не связан с дефицитом какого-либо витамина . Встречаются семейные случаи заболевания, однако генетическая роль в развитии эксфолиативного кератолиза изучена недостаточно [14] .
При обнаружении схожих симптомов проконсультируйтесь у врача. Не занимайтесь самолечением - это опасно для вашего здоровья!
Симптомы эксфолиативного кератолиза
Эксфолиативный кератолиз часто протекает без выраженных симптомов и проявляется незначительным поражением кожи ладонно-подошвенной области [5] . Отшелушиванию кожи предшествует появление наполненных воздухом пузырей, которые никогда не бывают заполнены жидкостью [3] . В некоторых случаях заболевание начинается только с очагового шелушения кожи, без образования пузырей.
После вскрытия пузырей остаются широкие сетчатые, круглые или овальные очаги. Они шелушатся, распространяются по периферии и образуют большие округлые участки, напоминающие кружева.
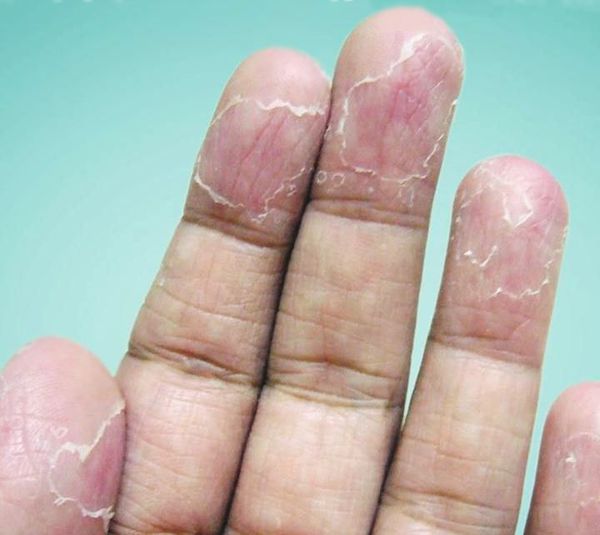
Участки слущенного эпителия теряют защитную функцию, становятся красными, сухими и покрываются трещинками. Шелушению может предшествовать небольшой зуд или жжение, в некоторых случаях область слущенного эпителия становится болезненной [3] .
Высыпания всегда симметричные. Иногда на кончиках пальцев образуются глубокие трещины, кожа становится жёсткой и немеет — в таком случае для полного заживления потребуется 1—3 недели. Эксфолиативный кератолиз может повториться через несколько недель после того, как на месте отшелушивания образовалась новая кожа.
Патогенез эксфолиативного кератолиза
Эпидермис — верхний наружный слой кожи, состоящий из кератиноцитов. Эти клетки содержат белок кератин, необходимый для прочности и эластичности кожи. Когда кератин разрушается, прочность кожи снижается, из-за чего она начинает шелушиться.
Также в эпидермисе содержатся корнеодесмосомы — белковые структуры, которые соединяют кератиноциты в сеть. В верхнем роговом слое кожи количество таких структур обычно уменьшается. Это нормальный физиологический процесс. Он называется десквамацией. Благодаря нему клетки кожи отшелушиваются, при этом поддерживается постоянная толщина рогового слоя [8] .
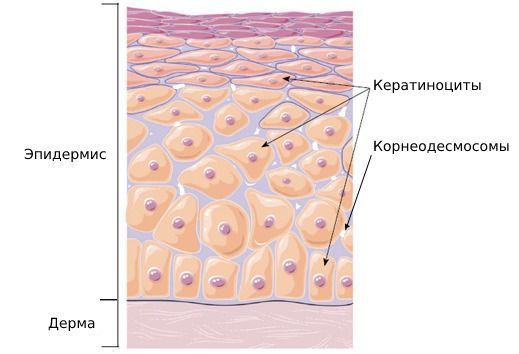
Исследование 24 пациентов с эксфолиативным кератолизом показало, что заболевание может возникать из-за дисбаланса активности ферментов, участвующих в процессе десквамации, особенно на коже ладоней [3] .
Точный механизм развития эксфолиативного кератолиза неизвестен. Прояснить возможные генетические или приобретённые причины заболевания помогут дальнейшие исследования десквамационных ферментов и ингибиторов — веществ, подавляющих или задерживающих течение ферментативных процессов. К таким веществам относятся ингибитор секреторной лейкоцитарной протеазы (SLPI), альфа-2 макроглобулин-1 (A2ML1), сульфат холестерина и ион цинка.
Классификация и стадии развития эксфолиативного кератолиза
По МКБ-10 (Международной классификации болезней) дерматологи часто кодируют эксфолиативный кератолиз как L26, относя заболевание к "другим эксфолиативным состояниям".
Классификации и стадийности эксфолиативный кератолиз не имеет. Заболевание иногда может приобретать хроническое течение с периодами ремиссии и обострения.
Осложнения эксфолиативного кератолиза
Эксфолиативный кератолиз не вызывает системных проявлений или осложнений. При заболевании может повреждаться кожа, в результате чего присоединяется бактериальная инфекция. Её признак — красные пятна, которые превращаются в гнойнички и пузырьки. Пузырьки безболезненные и легко вскрываются, образуются желтоватые чешуйки, так называемые "медовые корочки". При этом может возникать зуд.
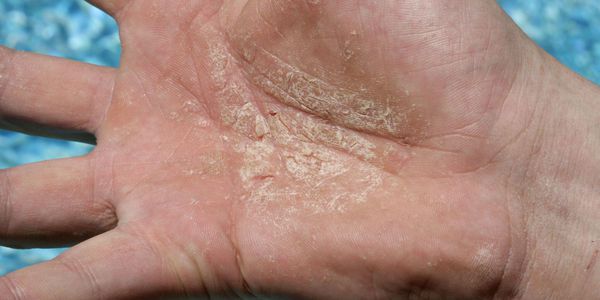
Диагностика эксфолиативного кератолиза
Диагноз "эксфолиативный кератолиз" ставится на основании данных клинического обследования и сбора анамнеза [15] . Некоторые пациенты отмечают, что состояние усугубляется после воздействия химических или физических раздражителей, таких как вода, мыло и моющие средства. Другие ассоциируют шелушение кожи с повышенным потоотделением.
Эксфолиативный кератолиз, вероятно, распространён, но часто протекает бессимптомно, поэтому врачи его наблюдают редко.
При осмотре отмечаются симметричные округлые участки шелушения на ладонях и, реже, на стопах. При этом воспаление на коже отсутствует.
Обычно дополнительное диагностическое тестирование не требуется. Однако в более сложных случаях, при подозрении на грибковое поражение, может потребоваться исследование с гидроксидом калия (KOH).
Биопсия кожи при кератолизе показывает расщепление и частично разрушенные корнеодесмосомы в роговом слое.
Патч-тесты , оценивающие потенциальную контактную аллергию , при эксфолиативном кератолизе отрицательны.
Дифференциальную диагностику проводят со следующими заболеваниями:
- различные формы дерматита рук, включая контактный дерматит, — для него характерен зуд, воздействие провоцирующих факторов в анамнезе, положительный ответ на гормональные мази;
- дисгидроз — сопровождается зудом, появлением трещин и везикул, наполненных жидкостью; — бляшки с чёткими границами бордово-красного цвета, выступающие над поверхностью кожи;
- дерматофития рук — изменение ногтей на руках и ногах, положительный ответ на противогрибковые препараты, не всегда симметричное поражение;
- простой буллёзный эпидермолиз — пузыри на разных участках кожи младенцев, возникающие после трения;
- ограниченный ладонный гипокератоз — редкое состояние, характеризуется центральной розовой областью с тонкой кожей на ладонях или подошвах стопы, по краям резкий переход к нормальной коже;
- пальмоплантарная кератодерма — возникает на коже стоп и кистей, характеризуется выраженным утолщением кожи [11] ;
- синдром акрального шелушения кожи — генетическое заболевание с пожизненным отслаиванием кожи.
Лечение эксфолиативного кератолиза
Причин возникновения эксфолиативного кератолиза может быть несколько, и не всегда они очевидны. Поэтому лечение болезни направлено на устранение симптомов и усугубляющих факторов. Это достигается защитой рук от физических или химических раздражителей ношением перчаток, когда это возможно.
Активное увлажнение кожи — важный, безопасный и эффективный метод лечения [10] . Наиболее подходящим способом для большинства пациентов являются кератолитические кремы, содержащие мочевину, молочную кислоту, лактат аммония или салициловую кислоту. Кремы с мочевиной увлажняют кожу и предотвращают её сухость. Кремы могут содержать мочевину 20 % или 40 %, 12 % лактата аммония, 6 % салициловой кислоты и 12 % молочной кислоты. Любой из них применяют до двух раз в день.

Приём наружных гормональных препаратов (стероидов) не требуется, так как воспаление отсутствует.
В некоторых исследованиях упоминается использование фотохимиотерапии с псораленом и ультрафиолетовым светом (PUVA), но только при тяжёлых случаях, так как риски этой терапии превышают пользу [9] .
PUVA-терапия заключается в приёме пациентом фотоактивного материала псоралена с последующим воздействием на кожу UVA лучей. Данных, подтверждающих пользу фототерапии при эксфолиативном кератолизе, на сегодняшний день недостаточно.
Возможные побочные эффекты PUVA-терапии: покраснение кожи вплоть образования пузырей, зуд; к редкими побочными эффектам относятся головная боль, головокружение, учащённое сердцебиение и слабость [4] .
Также в литературе встречаются данные о лечении эксфолиативного кератолиза ацитретином [7] [14] . Но для рутинного применения ацитретина в качестве лечения кератолиза информации пока недостаточно. Ацитретин — это производное витамина А, которым лечат псориаз.
Прогноз. Профилактика
Прогноз благоприятный. Обычно симптомы эксфолиативного кератолиза проходят самостоятельно или после прекращения контакта с провоцирующим фактором. Спустя несколько недель или месяцев формируется здоровая кожа. Однако через несколько недель может возникнуть рецидив.
Иногда кератолиз приобретает хронический характер и длится много лет подряд. В таком случае заболевание трудно поддаётся лечению. С возрастом эксфолиативный кератолиз возникает реже [2] .
Меры профилактики:
- избегать веществ, которые способствуют шелушению, например растворителей, антибактериального мыла и некоторых тканей;
- соблюдать гигиену рук и ног с частой стрижкой ногтей — это поможет избежать вторичного инфицирования;
- использовать перчатки при контакте с химическими веществами;
- наносить увлажняющие кремы для рук, особенно полезны кремы, содержащие мочевину, молочную кислоту или силикон.

Роль продуктов питания в развитии эксфолиативного кератолиза не доказана, поэтому соблюдать диету не нужно.
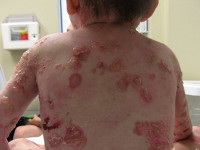
Буллезный эпидермолиз – группа наследственных заболеваний, которые характеризуются легкой ранимостью кожи, отсюда второе название этих патологий - «механобуллезная болезнь». Основным симптомом служит развитие на поверхности кожных покровов пузырей с серозным содержимым, после чего на их месте возникают долго незаживающие эрозии. Диагностика различных типов буллезного эпидермолиза осуществляется при помощи иммуногистологических и генетических методик, а также на основании данных осмотра пациента и изучения его наследственного анамнеза. Специфического лечения не существует, однако правильная и комплексная симптоматическая терапия может в ряде случаев значительно улучшать состояние больного.
МКБ-10
Общие сведения
Буллезный эпидермолиз – это гетерогенная группа наследственных заболеваний кожи, которые характеризуются образованием пузырей и эрозий в ответ на незначительное механическое воздействие. Впервые данный термин был использован в 1886 году немецким врачом-дерматологом Генрихом Кёбнером, дальнейшие исследования продемонстрировали, что существует множество разновидностей этой патологии. Генетические исследования буллезного эпидермолиза показали, что он может наследоваться как аутосомно-рецессивно, так и аутосомно-доминантно, с ним ассоциированы мутации более чем 10 генов. Существенные различия имеются и в клиническом течении разных типов этого заболевания, встречаемость колеблется в пределах 1:30000-1:1000000.
Патогенез нарушений при буллезном эпидермолизе долгое время оставался малоизученным. Прорыв в этом направлении произошел с внедрением в медицинскую практику электронной микроскопии, которая помогла визуализировать ультраструктуру пораженных тканей кожи. Следующий важный шаг в изучении буллезного эпидермолиза был совершен с открытием иммуногистологических исследований (иммунофлуоресценция). В настоящее время именно эти методики играют важнейшую роль в диагностике данных заболеваний, уступая по точности лишь генетическому анализу. Ввиду того, что методы изучения буллезного эпидермолиза постоянно совершенствовались, претерпевала изменения и классификация форм этой группы заболеваний.
Причины буллезного эпидермолиза
Этиология буллезного эпидермолиза неодинакова у разных типов заболевания, что в некоторых случаях достаточно сильно осложняет диагностику. Простой буллезный эпидермолиз обусловлен мутациями генов KRT5 и KRT14, однако, по данным врачей-генетиков, нарушением структуры этих генов объясняется только 75% случаев заболевания этого типа. При этом в кожных покровах, предположительно, нарушается равновесие в системе «ферменты-ингибиторы», и некоторые белки становятся объектом атаки. При простом буллезном эпидермолизе это могут быть протеины базальной мембраны (альфа6-бета4-интегрин) и белки десмосом базального слоя эпидермиса – десмоплакин, плакофиллин-1. В результате при механическом воздействии происходит выделение ферментов, которые разрушают указанные белки, тем самым провоцируя цитолиз и разрушение структуры эпидермиса, приводя к образованию пузырей.
Причиной развития другой формы патологии – пограничного буллезного эпидермолиза – являются мутации в генах LAMB3, LAMA3 и некоторых других. Большинство из этих мутации наследуется по аутосомно-рецессивному механизму, объектом атаки разбалансированной ферментной системы становятся такие протеины, как коллаген 17-го типа и ламинин-332. Эти белки участвуют в поддержании нормальной структуры нижних слоев эпидермиса, поэтому их повреждение приводит к характерным клиническим симптомам пограничного буллезного эпидермолиза. Помимо легкого образования пузырей и эрозий он характеризуется также повышенной ломкостью кожных покровов и более тяжелым течением.
Дистрофический тип буллезного эпидермолиза обусловлен мутациями в гене COL7A1, которые могут наследоваться как по аутосомно-доминантному, так и аутосомно-рецессивному механизмам. Белком-мишенью при этом выступает коллаген 7-го типа, который отвечает за стабильность структуры других соединительнотканных волокон кожи. Уменьшение количества этого протеина в тканях кожных покровов приводит к легкому развитию высыпаний, эрозий и пузырей, а также нередко сопровождается нарушениями других органов. В частности, дистрофический буллезный эпидермолиз часто приводит к развитию контрактуры суставов, поражение захватывает слизистые оболочки органов дыхательной и пищеварительной систем. На рубцах, которые остаются после заживления эрозий, нередко возникают злокачественные опухоли.
В целом, общий патогенез буллезного эпидермолиза можно свести к нарушению активности некоторых ферментов в тканях кожи. В результате этого разрушаются определенные ключевые структурные белки эпидермиса, дермы или базальной мембраны, что нарушает связи между клетками и приводит к образованию пузырей при механическом воздействии даже незначительной силы. Типы буллезного эпидермолиза отличаются один от другого локализацией пузырьков, видом мутации, что привела к этому заболеванию, и разновидностью белка, который стал объектом атаки ферментов.
Классификация буллезного эпидермолиза
В настоящий момент существуют десятки разновидностей буллезного эпидермолиза, которые достаточно трудно классифицировать в определенные группы. Проблема осложняется еще и тем, что почти за полтора века изучения данной патологии предпринимались неоднократные попытки разделить ее на определенные типы, используя самые современные на тот момент данные. В конечном итоге это привело к некоторой путанице, даже в научной литературе можно найти самые разнообразные варианты разделения буллезного эпидермолиза на разновидности. Наиболее современная классификация этого состояния в дерматологии включает в себя четыре типа заболевания, которые, в свою очередь, делятся на ряд подтипов:
- Простой буллезный эпидермолиз – имеет 12 подтипов, наиболее распространенными из которых являются синдромы Вебера-Коккейна, Кёбнера, Доулинга-Меары. Может наследоваться как аутосомно-доминантно, так и рецессивно, встречаемость составляет 1:100000. Простой буллезный эпидермолиз характеризуется образованием внутриэпидермальных или, реже, субэпидермальных пузырей, так как при этом заболевании поражаются белки эпидермиса.
- Пограничный буллезный эпидермолиз – делится на 2 подтипа, один из которых имеет еще 6 самостоятельных клинических форм. Наиболее тяжелой формой этого заболевания является подтип Херлитца, имеющий крайне высокую смертность. Встречаемость пограничного буллезного эпидермолиза составляет около 1:500000, образование пузырей при нем происходит на уровне светлой пластинки, что и дало ему название «пограничный».
- Дистрофический буллезный эпидермолиз – имеет два подтипа, которые делятся по механизму наследования этой патологии (доминантный и рецессивный подтипы). При этом встречаемость доминантного варианта несколько выше (3:1000000 против 1:500000 у рецессивной формы дистрофического буллезного эпидермолиза). Рецессивная разновидность также имеет несколько клинических форм, наиболее тяжелой из которых является подтип Аллопо-Сименса. При этом варианте заболевания у больных возникают глубокие эрозии, оставляющие после себя шрамы, возможны контрактуры суставов, поражение слизистых оболочек. Образование пузырей при этом происходит в сосочковом слое дермы, что и обуславливает появление шрамов и длительное заживление эрозий.
- Синдром Киндлера, или смешанный буллезный эпидермолиз, является одной из наиболее редких и малоизученных форм данной патологии. Особенностью, которая позволила выделить эту форму в отдельный тип, является образование пузырей во всех слоях кожи – эпидермисе, у светлой пластинке, в дерме. В настоящий момент определен только белок, выступающий в качестве мишени ферментов при смешанном буллезном эпидермолизе – киндлин-1.
Такой тип разделения всех клинических форм буллезного эпидермолиза является в настоящее время общепринятым. Но даже в пределах одного типа наблюдается большое разнообразие клинических симптомов заболевания, что осложняет диагностику и нередко влияет на прогноз патологии. Поэтому на сегодняшний день не прекращаются поиски более структурированной и приемлемой классификации буллезного эпидермолиза.
Симптомы буллезного эпидермолиза
Проявления буллезного эпидермолиза разных типов объединяет одно – развитие пузырей и эрозий в ответ на механическое воздействие на кожу. Различается лишь степень выраженности этих изменений, локализация, время существования и результаты заживления. При локализованной форме простого буллезного эпидермолиза (подтип Вебера-Коккейна) поражения располагаются только на определенном участке тела (руки, стопы). В младенческом возрасте возможна более широкая площадь появления пузырей, но с возрастом их выраженность уменьшается. Напротив, генерализованный подтип Доулинга-Меары характеризуется развитием мелких везикулярных высыпаний на значительной площади тела. Такой тип буллезного эпидермолиза возникает с самого раннего детства и может стать причиной смерти ребенка, итогом разрешения пузырьков может быть гиперкератоз, нарушения пигментации кожи, иногда возникает поражение слизистых.
Пограничная форма буллезного эпидермолиза протекает намного более тяжело, особенно так называемый летальный подтип Херлитца. При этом наблюдается повышенная ломкость кожных покровов, образование большого количества пузырьков, эрозий, на лице и спине часто возникают симметричные грануляции. Поражаются и слизистые оболочки рта, обнаруживается гипоплазия эмали и обусловленный ею тяжелый кариес. Столь тяжелое течение пограничного буллезного эпидермолиза часто становится причиной летального исхода в первые годы жизни. У выживших больных во взрослом возрасте формируются контрактуры суставов, поражение почек, потеря ногтей. Более легкая атрофическая форма пограничного буллезного эпидермолиза также характеризуется обширными высыпаниями, после разрешения которых формируются атрофические участки и рубцы. Также она часто приводит к дистрофии ногтей и рубцовой алопеции.
Дистрофический буллезный эпидермолиз практически всегда является генерализованным и поражает обширные участки тела. Доминантный вариант заболевания в целом отличается более доброкачественным течением, образование пузырей и их разрешение происходит медленно, однако большинство больных в конце концов теряют ногти на руках. После заживления эрозий на поверхности кожи формируются заметные рубцы. Рецессивный вариант дистрофического буллезного эпидермолиза, особенно его тяжелый генерализованный подтип, протекает намного тяжелее: помимо высыпаний у больных часто регистрируются псевдосиндактилии, обширные шрамы, потеря ногтей. Возникает поражение костей скелета, на месте заживших шрамов с годами может развиваться плоскоклеточный рак. Проблемой является еще и высокая устойчивость подтипа Аллопо-Сименса к терапевтическим мероприятиям.
Осложнения любого типа буллезного эпидермолиза сводятся к риску развития шока (при обширных поражениях), присоединения вторичной инфекции и спровоцированного ею сепсиса, обезвоживания больных. В большинстве случаев терапевтические процедуры производят только с целью недопущения этих состояний. Вероятность развития осложнений тем выше, чем большую область тела занимают патологические очаги и чем деструктивнее их характер (напряженные пузыри, эрозии, язвы).
Диагностика буллезного эпидермолиза
В настоящее время диагностика буллезного эпидермолиза осуществляется путем осмотра кожных покровов пациента, с помощью проведения иммуногистологических исследований и генетических анализов, в некоторых случаях производят изучение наследственного анамнеза. При осмотре кожных покровов специалист также может произвести диагностические тесты – механически воздействовать на кожу пациента и спустя время оценить результаты. Развитие на этом участке характерных для буллезного эпидермолиза пузырей или эрозий говорит в пользу наличия данного заболевания. На следующих этапах диагностики производят более точное определение формы патологии.
Иммунофлуоресцентный анализ при буллезном эпидермолизе осуществляется при помощи моно- и поликлональных антител, имеющих сродство к основным белкам эпидермиса, светлой пластинки и верхних слоев дермы. Это позволяет оценить количество того или иного белка, что, в свою очередь, говорит о ферментной активности тканей. Уменьшение количества того или иного белка свидетельствует о его низком выделении или же ускоренном разрушении. Снижение концентрации ключевых протеинов на определенных участках позволяет определить уровень развития пузырей на самом раннем этапе, что уже помогает с высокой долей вероятности определить тип буллезного эпидермолиза. Точку в диагностике этого состояния ставит генетический анализ методом прямого секвенирования генов, которые ассоциированы с тем или иным типом заболевания. Такой многостадийный подход к диагностике буллезного эпидермолиза обеспечивает высокую точность.
Значительно упростить диагностику этого заболевания позволяет изучение наследственного анамнеза пациента, по которому можно выявить его кровных родственников с такой же проблемой. Кроме того, если у кого-то из родных имеется буллезный эпидермолиз, имеет смысл производить пренатальную генетическую диагностику, что позволит выявить наличие данной патологии на ранних этапах развития плода. Дифференциальную диагностику осуществляют с истинной пузырчаткой, некоторыми формами буллезного пемфигоида, приобретенным буллезным эпидермолизом (который является не наследственным, а аутоиммунным заболеванием).
Лечение буллезного эпидермолиза
Специфического лечения этого заболевания не существует, все терапевтические процедуры сводятся к предупреждению развития осложнений и уменьшению выраженности пузырьков и эрозий. В случае тяжелых форм буллезного эпидермолиза назначают преднизолон. Из наружных терапевтических манипуляций производят асептическое вскрытие пузырьков, обработку их крышки антисептиками, накладывают гелиомициновую мазь. Наложение повязок нужно производить крайне осторожно, так как давление бинтов может спровоцировать появление новых пузырей. При наличии осложнений (шока, сепсиса) проводят симптоматическое лечение противошоковыми препаратами и антибиотиками. С профилактической целью можно производить облучение кожных покровов ультрафиолетовыми лучами.
Современная генетика и ряд других областей медицины продолжают широкие исследования буллезного эпидермолиза с целью поиска более эффективных методик лечения. Среди основных технологий и методов наиболее перспективными считаются способы с использованием стволовых клеток, белковая и генная терапии. Однако пока ни один из методов не вышел за рамки экспериментов на животных, поэтому буллезный эпидермолиз в настоящее время является неизлечимым заболеванием.
Прогноз буллезного эпидермолиза
Прогноз буллезного эпидермолиза чаще всего неопределенный, так как зависит от множества факторов и обстоятельств – типа заболевания, наличия или отсутствия у больного сопутствующих нарушений, его образа жизни. Например, локальный подтип простого эпидермолиза чаще всего имеет доброкачественное течение и редко создает угрозу жизни пациенту. Тогда как подтип Аллопо-Сименса имеет очень высокую смертность – как и от кожных проявлений, так и по причине отдаленных осложнений, таких как поражения почек и органов ЖКТ, а также развития плоскоклеточного рака кожи. Больные с такой проблемой должны бережно относиться к своей коже, не забывать про антисептическую обработку эрозий и других поражений, избегать занятий травмирующими видами спорта и иной деятельностью такого рода.
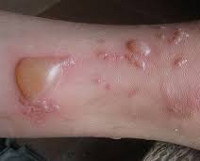
Буллёзный дерматит — это воспалительное поражение кожи с образованием на ней заполненных жидкостью пузырей. Чаще всего буллёзный дерматит возникает в результате контакта кожи с каким-либо агрессивным фактором внешней среды. Однако он может быть симптомом других дерматологических заболеваний, следствием метаболических и эндокринных нарушений или проявлением генетических аномалий. В диагностике буллёзного дерматита имеет большое значение определение воздействующего на кожу внешнего фактора, выявление сопутствующей патологии, лабораторная диагностики и биопсия.
Общие сведения
При буллёзном дерматите пузыри располагаются под эпидермисом (субэпидермально) или непосредственно в нем (интраэпидермально). Субэпидермальная локализация пузырей характерна для буллёзного пемфигоида, буллёзной формы системной красной волчанки, буллёзного эпидермолиза. Интраэпидермальные пузыри появляются при буллёзной эритродермии, болезни Хейли-Хейли. При размере более 5 мм пузыри называют буллами, менее 5 мм — везикулами.
Причины
К внешним факторам, вызывающим контактный буллёзный дерматит относится воздействие солнечных лучей и искусственных источников ультрафиолетового излучения, низких и высоких температур, агрессивных химических веществ (скипидар, урсол, краска для волос и т. п.), некоторых растений и лекарственных средств. При этом буллезный дерматит бывает обусловлен прямым воздействием фактора (простой контактный дерматит) или спровоцированной им аллергической реакцией (аллергический контактный дерматит).
Буллёзный дерматит может быть проявлением инфекционных заболеваний: герпес, буллёзная дерматофития, импетиго; воспалительных дерматозов: пузырчатка, буллёзная системная красная волчанка, буллёзный пемфигоид; метаболических нарушений: порфирия, пеллагра, диабетический буллёз, энтеропатический акродерматит; генетических аномалий: врожденная буллёзная эритродермия, болезнь Хейли-Хейли, буллёзный эпидермолиз.
Симптомы буллёзного дерматита
Буллёзный дерматит от воздействия низкой температуры — это отморожения. Они характеризуются первоначальным спазмом сосудов. Затем сосуды расширяются и на коже возникает покраснение, сопровождающееся чувством жжения и боли. Присоединяется отечность и появляются вялые пузыри с серозным или кровянистым содержимым. Эрозии, образующиеся после вскрытия пузырей, при заживлении покрываются корочками. Воздействие на кожу высокой температуры вызывает ожоги. Их клиническая картина сходна с отморожениями, но пузыри образуются сразу же после воздействия. Буллёзный дерматит возникает при отморожении или ожоге II степени.
Солнечный буллёзный дерматит развивается в течение нескольких часов после слишком длительного пребывания под прямыми лучами солнца. После покраснения кожи на ней образуются пузыри разного размера. При солнечном дерматите отмечается зуд, болезненность и жжение, возможно повышение температуры и нарушение общего самочувствия. После заживления эрозий на коже остаются участки гиперпигментации.
Буллёзный дерматит от химических факторов, возникая на участке кожи, контактировавшем с химическим веществом, может затем принимать генерализованный характер. Так, при контакте с урсолом излюбленное расположение пузырей при генерализации — это лицо и шея. Возникающий отек может захватывать веки с полным закрытием глазной щели.
Метаболический буллёзный дерматит развивается на фоне существующих эндокринных заболеваний или обменных нарушений. Диабетический буллёз возникает при сахарном диабете любого типа. При нем напряженные пузыри находятся на дистальных отделах ног или рук. Энтеропатический акродерматит связан с недостатком цинка и характеризуется локализацией пузырей на дистальных отделах конечностей, во рту, на губах и вокруг глаз.
Наследственный буллёзный дерматит обычно развивается сразу после рождения. Для буллёзного эпидермолиза характерно самопроизвольное внезапное появление пузырей и их образование в местах незначительного травмирования кожи. Болезнь Хейли-Хейли имеет клиническую картину пузырчатки, но передается наследственным путем.
Диагностика
В первую очередь проводится оценка клинической картины буллёзного дерматита. Имеет значение расположение пузырей, их характер, размер, количество и стадии развития, симметричность поражения, вовлечение слизистых оболочек.
В диагностике буллёзного дерматита контактной природы большое внимание уделяют выявлению провоцирующего фактора. При подозрении инфекционной природы пузырьков проводят бактериоскопию и посев содержащейся в них жидкости.
Одним из информативных методов диагностики буллёзного дерматита является биопсия с последующим гистологическим изучением. В качестве биопсийного материала берут свежий неповрежденный пузырь, немного захватывая расположенную вокруг него кожу. Для подтверждения аллергической природы дерматита в дополнение к гистологическом исследованию проводят реакции прямой и непрямой иммунофлуоресценции (РИФ).
Диагностику наследственных буллёзных дерматитов проводят с применением электронно-микроскопического исследования. При подозрении на порфирию исследуют мочу на наличие порфиринов, при подозрении на энтеропатический акродерматит в крови определяют концентрацию цинка.
Лечение буллёзного дерматита
В лечении контактного буллёзного дерматита основное — это устранение провоцирующего фактора. Если дерматит является проявлением или осложнением других заболеваний, то в первую очередь проводят лечение основного заболевания. При наследственных формах буллёзного дерматита проводится симптоматическое лечение.
Местно могут применяться кортикостероидные и антибактериальные мази, противовоспалительные смеси, средства способствующие заживлению эрозий, остающихся после пузырей. При необходимости производят вскрытие пузырей с тщательным соблюдением условий стерильности.
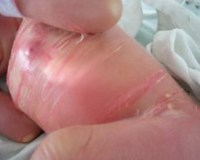
Буллезный ихтиоз — это крайне редкая форма генодерматоза, которая характеризуется избыточными процессами кератинизации, образованием многочисленных пузырей, желто-коричневых чешуек. Заболевание обусловлено аутосомно-доминантным типом наследования. Кожные изменения включают буллы, мокнущие эрозии, очаги гиперкератоза, ладонно-подошвенную кератодермию. Диагностика болезни предполагает исследование биоптатов кожи, молекулярно-генетические тесты. Методы лечения буллезного ихтиоза симптоматические: прием системных ретиноидов, наружная десквамативная терапия, аптечная косметика для ухода в домашних условиях.
МКБ-10
Общие сведения
Буллезный ихтиоз в медицинской литературе имеет множество синонимов: эпидермолитический ихтиоз, эпидермолитический гиперкератоз, буллезная врожденная ихтиозиформная эритродермия Брока. Заболевание было впервые описано русским дерматологом Никольским в 1897 г., а детальным изучением клинической картины патологии занимался французский ученый Л. Брок в начале ХХ века. Буллезная форма ихтиоза встречается с частотой 1 случай на 200-300 тыс. населения, половых и расовых различий в заболеваемости не обнаружено.
Причины
Ихтиоз с буллами может возникать при нескольких типах генных мутаций, наиболее распространенные – в генах KRT-1 (локус 12q13.3), KRT-10 (локус 17q21.2). Наследование генных дефектов происходит по аутосомно-доминантному типу с полной пенетрантностью, однако у 50% пациентов мутации происходят спорадически при неотягощенном семейном анамнезе. В современной генетике ведутся исследования для установления провоцирующих факторов спонтанных мутаций при генодерматозах.
Патогенез
Развитие буллезного ихтиоза связывают с повреждением генов, кодирующих образование кератина типа 1 (KRT-1) и типа 10 (KRT-10). Эти виды коллагеновых волокон в норме участвуют в поддержании структурной целостности супрабазальных кератиноцитов. Они отвечают за стабильность барьерной функции кожных покровов, формирование прочных межклеточных контактов, сохранение влаги в дерме.
Помимо типичных кожных симптомов, болезнь характеризуется множественными метаболическими расстройствами: изменениями липидограммы, протеинограммы, гиповитаминозами. Также нарушена работа кожных желез. В отличие от Х-сцепленного рецессивного ихтиоза, при буллезной форме не нарушается обмен стериновых протеаз, поэтому связи между ороговевшими клетками некрепкие, что обуславливает транссудацию жидкости и появление пузырей на коже.
Симптомы буллезного ихтиоза
Дерматологические проявления заболевания наблюдаются с рождения младенца. Кожные покровы новорожденного имеют ярко-розовую или красную окраску, они покрыты пузырями разного размера, заполненными прозрачной или мутноватой жидкостью. Характерная особенность этого варианта ихтиоза — участки неизмененной кожи выглядят естественно, что отличает состояние от синдрома «коллоидного плода», когда тело ребенка покрывается блестящей натянутой пленкой.
При давлении и трении легко формируются новые пузыри. При неосторожных прикосновениях буллы вскрываются, обнажая ярко-красные эрозивные мокнутия. Эрозии при буллезном ихтиозе очень медленно заживают, на их месте образуются застойные красные пятна, очаги шелушения. Вследствие чередования поверхностных ран, остатков отслоенного эпидермиса, крупных пузырей кожа ребенка напоминает обожженную.
Начиная с 2-3-летнего возраста отмечаются активные симптомы гиперкератоза. На теле появляются многочисленные чешуйки желтого или коричневого цвета, которые обычно окружены воспаленной покрасневшей кожей. Типичная локализация дерматологических элементов у страдающих буллезным ихтиозом — сгибательные поверхности конечностей, подмышечные впадины. Характерной особенностью заболевания является ладонно-подошвенная кератодермия.
Осложнения
Буллезный ихтиоз отличается тяжелым течением, не исключены летальные формы: аборты на ранних сроках, поздние выкидыши, рождение мертвого плода. Зачастую дети рождаются раньше срока, вследствие чего у них развивается респираторный дистресс-синдром («болезнь гиалиновых мембран»), внутримозговые кровоизлияния, ретинопатия недоношенных. Недоношенность может стать причиной некротического энтероколита, тяжелой гипогликемии, расстройств питания.
На фоне обширного поражения нарушаются все функции кожи, которые имеют первостепенное значение в периоде новорожденности — дыхательная, выделительная, терморегуляторная. Из-за массивной потери жидкости при вскрытии булл у младенцев возникает обезвоживание. Множественные открытые раны становятся входными воротами для инфекции, что в условиях недостаточного функционирования иммунитета новорожденного чревато септическими осложнениями.
Диагностика
Буллезный ихтиоз характеризуется типичными кожными проявлениями, поэтому предварительный диагноз ставится во время консультации детского дерматолога еще в первые недели жизни младенца. Для дифференциальной диагностики разных типов генодерматозов, исключения других вариантов дерматологических патологий, сопровождающихся образованием булл, пациентам назначаются такие методы исследования, как:
- Изучение кожной структуры. При электронной микроскопии биоптатов пораженных участков обнаруживаются изменения базальной пластинки эпидермиса, потеря контактов между клетками, липидоподобные вакуоли в клетках рогового и зернистого слоев.
- Анализы крови. Для уточнения тяжести состояния ребенка выполняется клиническое и биохимическое исследование крови. Чтобы исключить аллергический буллезный эпидермолиз, имеющий сходные кожные симптомы, рекомендована иммунограмма.
- Генетическое тестирование. Чтобы подтвердить наличие буллезного ихтиоза, производится секвенирование генома для поиска патогномоничных мутаций в локусах 17q21.2, 12q13.3.
- Пренатальная диагностика. В семьях, где есть больные буллезным ихтиозом, целесообразно проводить медико-генетическое консультирование беременных женщин. Для раннего выявления патологии по показаниям применяется амниоцентез или биопсия ворсин хориона.
Лечение буллезного ихтиоза
В период активного образования булл медицинская помощь пациенту сходна с протоколом ведения ожоговых больных. Для коррекции потерянной жидкости обеспечивается адекватная регидратационная терапия, для восполнения нутритивных потребностей ребенка используется парентеральное или зондовое питание. Обязательно применяется массивная антибиотикотерапия для профилактики осложнений инфекционного характера.
На этапе булл необходим бережный уход за ребенком, исключение травматизации кожи, ношение одежды и белья из максимально мягких тканей. Для уменьшения потерь жидкости накладываются повязки с белым вазелином, при обширном поражении эффективны гелевые компрессы из гидролизованного полимера. После появления признаков кератинизации схема терапии изменяется, назначается несколько групп препаратов:
- Кератолитики. Наружные составы с мочевиной, пропиленгликолем необходимы для размягчения очагов гиперкератоза, удаления ороговевших масс с поверхности кожи.
- Ретиноиды. Наружные и системные препараты рекомендованы взрослым пациентам для эффективного контроля гиперкератоза, стабилизации процессов кожной регенерации.
- Фруктовые кислоты. Лечебная косметика с кислотами подбирается для рутинного домашнего ухода, чтобы снизить активность кератинизации, вовремя отшелушивать роговые массы.
- Питательные кремы. Для предупреждения огрубения кожи, устранения сухости и дискомфорта наносится косметика, в состав которой входят глицерин, ланолин, растительные масла.
- Средства сSPF. Солнцезащитные кремы назначаются для регулярного применения, чтобы предотвратить избыточную пигментацию. SPF-средства являются обязательны компонентом ухода при лечении активными веществами (кислотами, ретиноидами).
Прогноз и профилактика
Буллезный ихтиоз является наиболее неблагоприятным вариантом среди всей группы нарушений кератинизации, поскольку он сопряжен с высоким уровнем антенатальной и младенческой смертности. Прогноз во многом зависит от объема буллезного поражения кожи, наличия инфекционных осложнений, своевременности и полноты медицинской помощи. Учитывая генетические предпосылки патологии и высокую частоту спонтанных мутаций, меры профилактики не разработаны.
1. Врожденный ихтиоз/ Т.С. Васильченко, А.А. Габдракипова// Вестник науки и образования. — 2020. — №24.
2. Ихтиоз: к вопросу наследования (обзор)/ Е.Е. Тальникова, В.Н. Шерстнева, А.В. Моррисон, С.Р. Утц// Саратовский научно-медицинский журнал. — 2016. — №3.
3. Принципы современного пересмотра классификации ихтиозов/ Л.Д. Калюжная// Клиническая иммунология, аллергология, инфектология. - 2015. — №1.
Читайте также: