
Гистиоцитоз кожи что это
Обновлено: 19.04.2024
Гистиоцитозы - классификация, диагностика, лечение
Гистиоцитозы - группа опухолевых заболеваний составляет 3-4% от всех опухолей детского возраста. Опухоли развиваются из макрофагально/моноцитарной группы клеток.
Эти клетки образуются в костном мозге, некоторое время циркулируют в крови в виде моноцитов, а затем мигрируют в ткани, где образуют макрофагальные элементы печени, селезенки, легких, костного мозга и тканей (гистиоциты) или антиген-продуцирующие клетки (дендритные клетки).
Заболевания, относящиеся к 1 классу, часто многоочагового характера, проявляют чувствительность к цитостатикам и излучению и могут приводить к смертельному исходу.
Классификация гистиоцитозов недавно была пересмотрена, и сейчас она основывается на характеристике инфильтрирующих гистиоцитов.
1. Гистиоцитозы 1 класса характеризуются присутствием клеток Лангерганса. Эти клетки могут быть незлокачественными и способны реагировать на медиаторы иммунного ответа. В электронном микроскопе в них можно видеть типичные гранулы Бирбека. Пока нет единого мнения по поводу того, является ли это заболевание злокачественным. Однако в некоторых исследованиях продемонстрирована моноклональная природа этих клеток.
2. Гистиоцитозы класса 2 характеризуются наличием инфильтратов реактивных макрофагов. К этому классу относятся два редких вида заболевания, которые быстро заканчиваются смертельным исходом, семейный эритрофагоцитарный лимфогистиоцитоз и инфекционный гемофагоцитарный синдром.
3. К классу 3 относятся злокачественные опухоли гистиоцитарного происхождения, включая моноцитарный лейкоз.

Гистиоцитоз из клеток Лангерганса (1 класс гистиоцитозов)
Гистиоцитоз из клеток Лангерганса (ГКЛ) обычно называют гистиоцитозом X. Он объединяет целый ряд заболеваний: болезнь Леттерера-Сиве эозинофильную гранулему и болезнь Хенда-Шюллера-Кристиана. Болезнь Леттерера-Сиве представляет собой острое заболевание детского возраста, характеризующееся гепатоспленомегалией, набуханием лимфатических узлов, тромбоцитопенией и развитием кожной сыпи.
При обследовании обычно обнаруживаются себор-рейные высыпания на волосистой части головы, имеющие вид чешуйчатых напластований Образование многочисленных инфильтратов в коже, печени, селезенке и в костном мозге приводит к фунциональным расстройствам печени, одышке и к нарушениям функции костного мозга. Часто в костях развиваются литические очаги. Болезнь, ранее известная под названием эозинофильная гранулема, характеризуется рядом синдромов: от отдельных повреждений, развивающихся на костях, обычно у старших детей, до множественных проникающих костных повреждений.
Для детей моложе 3 лет прогноз оказывается неблагоприятным. Обычно детей беспокоят боли в костях, при осмотре обнаруживается лимфоаденопатия. Известны случаи спонтанного излечения. У некоторых детей обнаруживаются множественные эозинофильные гранулемы.
Иногда диагносцируется несахарный диабет, который развивается на почве поражения гипофиза или вызывается опухолью локализованной в глазнице (болезнь Хенда-Шюллера-Кристиана). Также наблюдается увеличение размеров печени и селезенки и появление кожных инфильтратов, однако эта болезнь носит менее агрессивный характер, чем болезнь Леттерера-Сиве. Как при эозинофильной гранулеме, развитие болезни может прекратиться, и по прошествии 3-4 лет рецидивы больше не наблюдаются.
Несахарный диабет принимает хроническую форму. Может развиться такое хроническое неврологическое расстройство как печеночная энцефалопатия на фоне цирроза с портальной гипертензией.
Прогноз и лечение гистиоцитозов у детей
Максимальная смертность наблюдается у маленьких детей и при инфильтрации опухолью внутренних органов (легких, печени и костного мозга). Отдельные повреждения костей лечат выскабливанием и местным назначением стероидов. Нередко эффективной оказывается лучевая терапия, назначенная в небольших дозах, однако при этом увеличивается риск развития костных сарком в отдаленные сроки после облучения. Больных с более распространенным опухолевым процессом можно лечить цитостатиками.
Наиболее часто назначают преднизолон, винбластин, этопозид, хлорамбуцил и метотрексат. Однако болезнь может протекать очень вяло и проходить даже без лечения, поэтому крайне важно избегать развития токсикозов. Во многих случаях (особенно когда болезнь ограничена поражением костей) лечение можно приостановить на длительный срок.
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
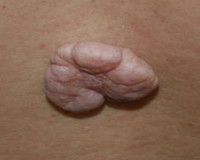
Ретикулогистиоцитома кожи – доброкачественная гигантоклеточная гранулёма фиброгистиоцитарного типа, опухоль соединительной ткани. Характеризуется бурыми узловатыми опухолевидными высыпаниями размером от жемчужины до перепелиного яйца, безболезненными, иногда немного зудящими, возвышающимися над уровнем здоровой кожи. Узлы имеют тенденцию к слиянию и группировке или распространяются по всей поверхности кожи, иногда самостоятельно инволютируют, оставляя атрофические изменения. Диагностируют опухоль клинически с учётом результатов биопсии. Специальная терапия обычно не требуется, иногда выполняют хирургическое удаление или применяют кортикостероиды в сочетании с цитостатиками.
Общие сведения
Ретикулогистиоцитома кожи – доброкачественная очаговая (солитарная или множественная) дисплазия ретикулярной ткани неясного генеза, реактивный гистиоцитоз. Патологический процесс не имеет расовых и сезонных особенностей, не обладает эндемичностью. Чаще поражает женщин зрелого возраста, нередко развивается на фоне туберкулёза, заболеванию может предшествовать системная полиартропатия. По данным зарубежных исследователей, в 27% случаев при ретикулогистиоцитоме кожи диагностируют рак внутренних органов, при этом кожная опухоль склонна к спонтанной инволюции. В отечественной дерматологии первым описал ретикулогистиоцитому (фиброму) в 1902 году знаменитый московский врач Е.С. Главче.
Западные дерматологические школы рассматривают патологический процесс, как реактивно-воспалительный гранулематоз, редкое проявление малигнитета (злокачественности), особую онконозологию. По данным отечественных авторов, ретикулогистиоцитоз ассоциирован с опухолями в 10-25% случаев, что и определяет актуальность проблемы на современном этапе.
Причины ретикулогистиоцитомы кожи
Сущность патологического процесса неясна. Есть разные взгляды на природу возникновения ретикулогистиоцитомы. Одни дерматологи относят неоплазию к реактивному гранулематозу, ангиофиброксантоме, другие – к дисплазии ретикулярной ткани, но большинство рассматривают ретикулогистиоцитому в качестве самостоятельной нозологии неясного генеза. При этом в механизме развития патологии приоритет отдают изменениям в фагоцитарных гистиоцитах, связанных с ними нарушениях в формировании и развитии соединительной ткани. По сути, дерма теряет стабильность, возникают аномалии прочности коллагеновых структур, визуально проявляющиеся той или иной патологией кожи.
Экзогенные и эндогенные триггеры, к которым, вероятнее всего, относятся генетические мутации, инфекции, системные заболевания, экологическая обстановка, нерациональное питание и стресс, дестабилизируют постоянство внутренней среды, что ведёт к внутрикожному дисбалансу, являющемуся пусковым моментом в развитии аутоиммунных и аллергических реакций. При этом нарушенная адаптационная способность кожи стимулирует формирование разных проявлений дермальной недостаточности.
Параллельно теряется способность к восстановлению нарушенной микроциркуляции дермы и её регенерации, происходит разбалансировка синтеза коллагена. Коллагеновые волокна меняет коллагеназа, синтезируемая фибробластами. Деградация коллагена активирует гистиоцитарные макрофаги и цитокины, стимулирующие воспаление в соединительной ткани и пролиферацию клеток кожи. Поскольку в дерме соединительнотканная основа и русло микроциркуляции объединены в одну трофико-транспортную систему, нарушается лимфообращение и кровоток, что визуально проявляется интерстициальным отёком с исходом в фиброз.
Можно расценивать происходящие изменения не в качестве заболевания, а в качестве определённого состояния – ответа кожи на неблагоприятные факторы, считать это особым переходным состоянием от здоровья к нездоровью. Однако нельзя исключить и формирование самостоятельной нозологии с вовлечением в процесс многих внутренних органов. При ретикулогистиоцитоме кожи встречаются опухоли пищеварительной системы, бронхов, матки и её придатков, молочной железы, а так же лимфома, меланома и миеломная болезнь.
Классификация и симптомы ретикулогистиоцитомы кожи
Классификации данного патологического процесса в дерматологии не существует, но для понимания сути ретикулогистиоцитомы кожи необходимо учитывать, что по Международной гистологической классификации ВОЗ она включена в группу опухолей соединительной ткани наряду с ксантомами, фибромами, атипичными фиброксантомами и ювенильными ксантогранулемами.
Заболевание начинается спонтанно, появляются очаги поражения кожи, состоящие из плоских или элипсоидоподобных бурых безболезненных узлов различного размера (от ягод рябины до перепелиного яйца), слегка возвышающихся над уровнем здоровой кожи. Высыпание первичных элементов сопровождается неинтенсивным зудом, со временем узлы приобретают синюшный оттенок. Некоторые плоские образования заметны при осмотре только из-за их интенсивной окраски. Опухолевидные очаги имеют плотноэластическую консистенцию, сливаясь, образуют бугристые конгломераты значительных размеров. Первичные элементы ретикулогистиоцитомы кожи могут существовать солитарно, но встречаются и множественные опухоли, локализующиеся на разных участках кожного покрова (волосистая часть кожи головы, лицо, спина, грудь, конечности, область половых органов).
В отдельных случаях мелкие узелки располагаются вокруг ногтевого валика в виде кораллового ожерелья, результатом патологического процесса становится дистрофия ногтя. Описаны случаи кистозного поражения сухожилий крупными узлами ретикулогистиоцитомы. В 50% случаев к поражению кожи присоединяются высыпания на слизистых оболочках полости рта, верхних дыхательных путей, глотки, языка и губ. Иногда опухолевидные образования приобретают способность к распространению или начинают группироваться в каком-то одном регионе, практически наползая друг на друга. Очаги ретикулогистиоцитомы кожи обладают медленным ростом, при этом узлы в некоторых случаях спонтанно инволюционируют без всяких видимых причин, оставляя выраженные атрофически-пигментные изменения на коже.
Самой серьёзной сопутствующей патологией ретикулогистиоцитомы кожи является поражение суставов, которое предшествует визуальным проявлениям патологического процесса. С симптомами симметричного деформирующего полиартрита к врачам обращаются около 60% больных. Стоить отметить, что во многих случаях ретикулогистиоцитомы кожи возникают на фоне латентно текущей туберкулёзной или онкологической патологии, при этом пациент прогрессивно теряет вес. Описаны поражения мышц, лимфоузлов, костного мозга и практически всех внутренних органов. Вовлечение сердца в патологический процесс приводят к летальному исходу.
Диагностика и лечение ретикулогистиоцитомы кожи
Клинический диагноз ставит дерматолог на основании симптомов, полного клинико-лабораторного обследования и данных биопсии накожных элементов. Дифференцируют ретикулогистиоцитому кожи с ревматоидным артритом, ретикулосаркоматозом кожи, саркоидозом, подагрой, ангиофиброксантомой, ретикулогистиоцитозом, дерматофибромой, фибросаркомой, ювенильной ксантогранулемой. Обычно опухоль не требует специального лечения, иногда после консультации с хирургом солитарные узлы удаляют хирургическим путем. Иногда хороший эффект дает лечение кортикостероидами в сочетании с цитостатиками. При отсутствии фоновых заболеваний прогноз достаточно благоприятный, при наличии такой патологии исход зависит тяжести и вида фонового процесса.
Гемофагоцитарный лимфогистиоцитоз – группа врожденных и приобретенных заболеваний, возникающих вследствие нарушений регуляции иммунного ответа и характеризующихся гиперпродукцией гистиоцитов, а также цитотоксических T-лимфоцитов. Клиническая симптоматика проявляется фебрильной лихорадкой, увеличением печени и селезенки, периферических лимфатических узлов, поражением нервной системы, костного мозга и других органов. Диагностика основана на данных клинического, лабораторного (цитопения, коагулопатия, билирубинемия и др.) и инструментального обследования. Лечение: иммуносупрессивная терапия, кортикостероиды, воздействие на причинные факторы.
МКБ-10
Общие сведения
Гемофагоцитарный лимфогистиоцитоз (гемофагоцитарный синдром) – врожденное или приобретенное нарушение регуляции иммунного ответа, при котором происходит аномальная активация цитотоксических T-лимфоцитов, моноцитов и макрофагов с аккумуляцией в органах-мишенях и развитием в них выраженного патологического процесса (воспаления, повреждения тканей, фагоцитоза форменных элементов крови). При врожденной, генетически детерминированной форме заболевания болеют преимущественно дети раннего возраста и в 60-80% случаев – на первом году жизни.
Вторичный (приобретенный) гемофагоцитарный лимфогистиоцитоз встречается во всех возрастных категориях, развивается на фоне затяжного течения различных инфекционных заболеваний, аутоиммунных процессов и новообразований. Впервые признаки гемофагоцитарного синдрома были описаны еще в 1939 году, а заболевание тогда было названо гистиоцитарным медуллярным ретикулезом. Семейная наследственная форма лимфогистиоцитоза была впервые описана в 1959 году. Распространенность заболевания колеблется от 1 случая на 50 тысяч новорожденных до 1-2 случаев на 1 миллион детей в возрасте до 15 лет.
Причины гемофагоцитарного лимфогистиоцитоза
Врожденный гемофагоцитарный лимфогистиоцитоз возникает вследствие генетического дефекта механизмов клеточной цитотоксичности из-за мутаций гена перфорина. В норме регуляция иммунного ответа обеспечивается своевременным ограничением активности эффекторов иммунной системы в процессе ликвидации угрозы для организма со стороны проникших инфекционных агентов и других негативных воздействий. В этой регуляции важную роль играют механизмы клеточной цитотоксичности.
При первичном гемофагоцитарном лимфогистиоцитозе регулятивная роль цитотоксических гранул клеток и цитотоксических T-лимфоцитов в отношении клеток-мишеней нарушается, происходит чрезмерная активация иммунных клеток, в избытке продуцируются провоспалительные цитокины (интерфероны, фактор некроза опухоли и др.). Активированные вследствие «цитокинового шторма» T-лимфоциты и макрофаги инфильтрируют, а затем повреждают органы и ткани. Важным фактором патогенеза заболевания является развитие патологического гемофагоцитоза зрелых форменных элементов крови. Такой фагоцитоз происходит и в норме, помогая организму избавиться от старых клеток. В данном же случае макрофаги начинают фагоцитировать полноценные, нормально функционирующие форменные элементы крови, приводя к выраженной цитопении, коагулопатии и другим проявлениям заболевания.
Приобретенный (вторичный) гемофагоцитарный лимфогистиоцитоз развивается на фоне некоторых инфекционных заболеваний, опухолей, аутоиммунных процессов, при трансплантации органов и тканей, причем характерные нарушения регуляции иммунного ответа могут быть как следствием основного заболевания, так и осложнением, связанным с проведением иммуносупрессивной терапии и возникновением вторичной инфекции.
Симптомы гемофагоцитарного лимфогистиоцитоза
Клинические проявления гемофагоцитарного лимфогистиоцитоза чрезвычайно вариабельны. Наиболее часто наблюдается длительная лихорадка, рефрактерная к проводимой антибактериальной и противовирусной терапии. Озноб, признаки общей интоксикации (слабость, потливость, нарушения сна, отказ от приема пищи, тошнота и рвота, боли в мышцах) продолжаются в течение длительного времени, приобретая волнообразное течение с периодическими временными улучшениями самочувствия.
Характерным проявлением заболевания является увеличение печени и селезенки, имеющее прогрессирующий характер. К ранним симптомам первичного гемофагоцитарного лимфогистиоцитоза относится появление кожной сыпи, увеличение периферических лимфатических узлов, а также развитие неврологических расстройств в виде повышенной возбудимости, двигательных нарушений и расстройств чувствительности, судорожного синдрома, признаков повышения внутричерепного давления у детей раннего возраста. Встречаются при гемофагоцитарном лимфогистиоцитозе и симптомы, свидетельствующие о наличии анемии, коагулопатии – бледность и желтушность кожных покровов, периферические отеки, признаки кровотечения из пищеварительного тракта (черный кал, примесь крови в каловых массах и др.).
При вторичном гемофагоцитарном синдроме сочетаются клинические признаки поражения иммунной системы и проявления основного заболевания (вирусной инфекции, злокачественного новообразования, аутоиммунного заболевания).
Диагностика гемофагоцитарного лимфогистиоцитоза
Диагноз устанавливается в результате тщательного изучения анамнеза, клинической картины заболевания, результатов лабораторных и инструментальных исследований. Необходимы осмотры врача-гематолога, аллерголога-иммунолога, онколога, инфекциониста, ревматолога и других специалистов. Разработаны международные диагностические критерии гемофагоцитарного лимфогистиоцитоза, к которым относятся:
- лихорадка с повышением температуры выше 38,5 градусов, продолжающаяся более недели;
- увеличение печени и селезенки;
- цитопения с уменьшением гемоглобина ниже 90 г/л, тромбоцитов – меньше 100000 клеток/мкл, нейтрофилов – меньше 1000/мкл;
- признаки коагулопатии;
- увеличение ферритина больше 500 нг/мл;
- повышение уровня растворимого sCD25 в крови;
- низкое или полное отсутствие активности NK-клеток.
Приобретенные формы гемофагоцитарного синдрома диагностируются на основании вышеописанных международных критериев и проведения уточненной диагностики основного заболевания, для чего зачастую необходимо выполнять сложные лабораторные и инструментальные исследования (эндоскопические, УЗИ, КТ, МРТ, ПЭТ).
Дифференциальный диагноз гемофагоцитарного лимфогистиоцитоза проводится с:
- различными врожденными и приобретенными иммунными заболеваниями,
- болезнями крови,
- острыми и хроническими вирусными инфекциями,
- злокачественными новообразованиями (острым лимфобластным лейкозом, неходжкинскими лимфомами, другими злокачественными опухолями после проведенной химиотерапии),
- системными заболеваниями соединительной ткани: системной красной волчанкой, ювенильным дерматомиозитом, узелковым периартериитом, ювенильным ревматоидным артритом.
Лечение гемофагоцитарного лимфогистиоцитоза
Современная тактика лечения наследственной формы гемофагоцитарного лимфогистиоцитоза включает проведение химиотерапии с использованием иммуносупрессивных средств (дексаметазона, этопозида, циклоспорина A), а также трансплантации стволовых клеток. Прогноз заболевания значительно улучшается при своевременном проведении трансплантации от гистосовместимого родственного донора.
При вирусных, бактериальных и паразитарных инфекциях, вызвавших появление гемофагоцитарного синдрома, проводится этиотропная антимикробная терапия, инфузии высокодозного иммуноглобулина, а также иммуносупрессивная терапия с введением циклоспорина A и кортикостероидов. Иногда показано проведение трансплантации костного мозга. При опухолях, наряду с лечением основного заболевания, в терапевтическую схему включаются иммуносупрессивные средства в индивидуально подобранных дозах. При аутоиммунных процессах лечение включает сочетание иммуноглобулина, пульс-терапии с использованием кортикостероидов, а также циклоспорина A.

Врач ультразвуковой диагностики. Стаж 20+ лет. Принимает в Университетской клинике в Санкт-Петербурге. Стоимость приема 1100 руб.
- Запись опубликована: 15.06.2022
- Reading time: 4 минут чтения
Саркоидоз — это иммунное заболевание, возникающее в результате воспаления тканей. Патология может развиваться в любом органе, но обычно начинается в легких или лимфатических узлах. Процесс может затрагивать печень, кожу, сердце, нервную систему и почки. Характерная особенность саркоидоза — образование гранулем. В большинстве случаев эти изменения проходят спонтанно. Если этого не происходит, за счет воспаления образуется фиброз.
Вероятная причина саркоидоза — действие специфического фактора окружающей среды (фактора саркоидоза) на иммунную систему генетически предрасположенных людей, но этот фактор еще не обнаружен. Можно было только приблизительно оценить его свойства.
Что такое саркоидоз?
Саркоидоз – это воспалительное заболевание, характеризующееся образованием гранулем – скоплений воспалительных клеток – в одном или нескольких органах тела. Когда иммунитет слишком активен и образуется много гранулем, они нарушают структуру и мешают функциям органа. Неконтролируемое хроническое воспаление приводит к фиброзу, представляющему собой постоянное рубцевание ткани органа.
Специфика и тяжесть заболевания варьируются. Заболевание может развиваться внезапно и так же быстро исчезать. Иногда оно развивается постепенно, и симптомы рецидивируют на протяжении всей жизни. Трети больных требуется длительное лечение. Саркоидоз считается хроническим, если заболевание остается активным более 2-х лет.
Саркоидоз был впервые описан в 1877 году английским дерматологом Джонатаном Хатчинсоном, обнаружившим у больного многочисленные странные поражения кожи конечностей. Термин «саркоид» ввел чуть позже ученый Бек. Название болезни происходит от греческих слов сарк и оид., или «похожий на мясо». Этот термин относится к кожному саркоидозу, часто сопровождающемуся некрасивыми высыпаниями. Д. Хатчинсон также описал изменения в легких, лимфатических узлах, слюнных железах, костях, селезенке и слизистых оболочках.
Многоорганная природа этого заболевания, с особым упором на поражение легких и лимфатической системы, была хорошо раскрыта уч. Шауманом (1917), связавшего поражения кожи и органов в единое системное заболевание. Как и многие другие ученые, Шауман считал саркоидоз одной из форм туберкулеза. Термин «саркоидоз» был официально принят только в 1960-х годах, и болезнь стала называться болезнью Бенье-Бека-Шаумана (BBS).
Ещё одно важное открытие в этой области было сделано ученым С. Лефгреном в 1946 г. Он обнаружил, что на ранних стадиях саркоидоза может наблюдаться двустороннее увеличение лимфоузлов, узловатая эритема, повышение температуры тела и изменения в крупных суставах. Комплекс этих симптомов получил название синдрома Лефгрена.
Распространенность патологии
Саркоидоз — наиболее распространенное интерстициальное заболевание легких с ежегодной заболеваемостью в мире от 1 до 64 случаев на 100000 жителей. Максимальная заболеваемость наблюдается в странах Северной Европы и среди афроамериканцев в США, а самая низкая — в Сингапуре и Японии.
Статистика показывает, что патология затрагивает разные органы в зависимости от национальности или скорее места жительства: у людей с темной кожей часто наблюдается кожный саркаидоз, у японцев страдает сердце и нервная система.
В России саркоидоз также встречается даже в тяжелой форме. По статистике, от него умирает почти пол процента всех больных. Причины гибели — сердечно-лёгочная недостаточность, нейро- и кардиосаркоидоз.
Патология чаще всего возникает в возрасте от 20 до 40 лет. Более подвержены этому заболеванию женщины. Увеличивает риск возникновения патологии семейный анамнез.
Причины развития саркоидоза — теория и факты
Некоторые исследования показывают, что могут вызывать заболевание бактерии, вирусы или химические вещества. Такие триггеры, хотя обычно безвредны для большинства людей, могут раздражать иммунную систему людей с генетическим риском развития саркоидоза.
Существуют теории, что иммунный ответ на раздражитель может быть слишком активным или даже неуместным, что приводит к воспалению, образованию гранулемы, а в некоторых случаях рубцеванию или фиброзу. Появляется все больше доказательств того, что этот иммунный ответ на саркоидоз может также включать аутоиммунный ответ на «собственные» белки. Однако пока саркоидоз не считается преимущественно аутоиммунным заболеванием, как например, ревматоидный артрит или системная красная волчанка, хотя исследования и показывают, что некоторые иммунные реакции и генетические факторы у этих патологий схожи.
На самом деле, что именно вызывает саркоидоз, доподлинно неизвестно. Поэтому правильно говорить, что саркоидоз связан с действием саркоидного фактора окружающей среды на иммунную систему генетически предрасположенного человека. Сам фактор, запускающий иммунный ответ, пока не выявлен, поэтому его свойства можно оценить только приблизительно.
Саркоидозный фактор характеризуется плохими растворимостью и разлагаемостью внутриклеточными ферментами, кислотами, под воздействием высокой температуры, органических растворителей, нейтральных детергентов. Этот факт указывает, что речь идет о белке, способном выживать внутри макрофагов, вызывая хроническую стимуляцию.
Саркоидозный фактор характеризуется иммуногенностью, то есть способностью индуцировать олигоклональную пролиферацию CD + Т-клеток. Именно этот фактор является промотором (по крайней мере, в начальной фазе заболевания) воспаления с преобладанием Th1-лимфоцитов. Это приводит к увеличению активности цитокинов: IFN-γ, IL-2, TNF-α. Этот же фактор с участием Т-лимфоцитов, в основном CD+ стимулирует образование грануляционной ткани, состоящей из эпителиальных и гигантских клеток.
Предрасположенность к саркоидозу увеличивает наличие аллелей: HLA-DR3, HLAS-DR5, HLA-DR8, HLA-DR9, HLA-DR12, HLA-DR14, HLA-DR15, HLA-DR17, HLA-DPB1, HLA-DQB1. Известны также аллели, отвечающие за защиту: HLA-DR1, HLA-DR4.
Исследования связи молекул HLA с возникновением и течением саркоидоза показали, что в скандинавской популяции гаплотип HLA-DR17 связан с острой формой саркоидоза, а HLA-DR14 и HLA-DR15 — с хронической. Внелегочная форма у итальянского населения связана с гаплотипом HLA-B22.
Интересные факты дало и американское исследование ACCES с участием более 1400 человек. Ученые не нашли прямой связи между саркоидозом и инфекционными агентами: HHV-8, HCV, CMV, ретровирусом, вирусом Коксаки B и EBV. При этом они выяснили, что саркоидоз часто развивается у людей, подвергающихся воздействию пыли, газов и органических частиц.
В 1940-х годах было обнаружено, что сходные с саркоидозом симптомы дает хроническая гранулематозная болезнь (ХГБ), вызываемая воздействием металла бериллия. Для дифференциации этих заболеваний проводится тест на бласт-трансформацию лимфоцитов в присутствии соли бериллия.
К группам профессионального риска по ХГБ относятся: зубные техники, ювелиры, рабочие, занятые в процессах восстановления металлов и в оружейной промышленности. Саркоидоз чаще встречается у пожарных. Этиологический фактор — токсины, выделяющиеся при высоких температурах.
Группы риска
Наиболее высокие риски заболеть саркоидозом у следующих групп пациентов:
- Подвергшиеся инфекциям. Бактериальным, вызванным микобактериями, Propionibacterium acnes, возбудителем болезни Лайма. Вирусным — гепатиту С, герпесвирусу, вирусу Д. Каннингема.
- Находящиеся в загрязненной среде. Опасно воздействие микрочастиц металлов. Особенно часто саркоидоз связывают с бериллием, медью, алюминием, барием, лантаноидами, кобальтом, золотом, цирконием, титаном. Также опасна любая пыль, плесень, дым.
- Имеющие родственников с саркаидозом.
- Курящие. Саркоидоз у таких больных часто протекает в тяжелой форме.
Механизм образования гранулем при саркоидозе
Ранний саркоидоз характеризуется накоплением большого количества активированных макрофагов и CD4+ Т-клеток в легких. Так же образуются и внелегочные очаги с воспалительным и иммунным процессами. Лимфоциты Th1 продуцируют гамма-интерферон (IFN-γ), интерлейкин 2 (IL-2) и другие цитокины, запускающие воспаление.
Важную роль в воспалении при саркоидозе играют макрофаги — источник провоспалительных цитокинов TNF-α, IL-12, IL-17 и факторов роста. Это постоянная часть саркоидных гранулем, состоящих из эпителиальных клеток, окруженных неравномерным слоем Т- и В-лимфоцитов. CD4+-лимфоциты доминируют во всех участках, где образуются гранулемы. Соотношение лимфоцитов CD4+ к CD8+ может быть более 10.
По мере развития фибробласты они размножаются и производят коллаген, заменяющий всю опухоль. Иногда в узелке можно найти две микроскопические структуры: микроскопические астероидные тела внутри гигантских клеток и тела Шаумана – слоистые плотности, состоящие из кальция и белков. Эти структуры бесполезны в диагностике саркоидоза, так как они также присутствуют в гранулемах другого происхождения.
При саркоидозе иногда можно обнаружить центральный некроз, указывающий на воспалительный процесс. Серозный некроз, типичный для туберкулеза, при саркоидозе отсутствует. Воспаление происходит из-за перераспределения клеток из периферической крови в легкие и локальной пролиферации этих клеток.
Цитокины, ответственные за привлечение клеток к тканям: IL-8, IL-15, IL-16 и RANTES (хемотаксис лимфоцитов), синтезируемый Т-лимфоцитами. С другой стороны, IL-2 отвечает за пролиферацию клеток, действуя как местный фактор роста для Т-лимфоцитов, инфильтрирующих паренхиму легких и другие ткани, в которых происходит саркоидозная реакция.
В воспалительную саркоидную реакцию вовлечены четыре типа цитокинов:
- цитокины из лимфоцитов Th1: IFN-γ, IL-2, IL-12, IL-15, IL-18, IL-27;
- провоспалительные цитокины: TNF-α, IL-1, GM-CSF, IL-62;
- противовоспалительные цитокины: ИЛ-10, трансформирующий фактор роста β (TGF-β);
- профибротические цитокины: TGF-β, фактор роста тромбоцитов (PDGF), инсулиноподобный фактор роста 1 (IGF-1).
Наиболее важные клетки, определяющие развитие саркоидозного воспаления, — лимфоциты Th1. Процесс поражает многие органы и ткани, но в 90% случаев это легкие. Т-лимфоциты находятся в интерстициальной ткани и в воздушных пространствах.
Саркоидозные гранулемы образуются в ответ на постоянный антигенный раздражитель. Фазы формирования гранулем:
- Вовлечение Th1-лимфоцитов с фенотипом CD4+ в результате действия антигенпрезентирующих клеток;
- Секреция провоспалительных и профибротических цитокинов;
- Скопление иммунокомпетентных клеток в очагах воспаления, то есть в легких, лимфатических узлах, коже, печени, селезенке, слюнных железах, сердце, нервной системе, мышцах.
В первой фазе саркоидозного воспаления процесс обратим, потому что процессы фибриногенеза подавлены. Т-клетки устойчивы к апоптозу (запрограммированная гибель клеток) и накапливаются в тканях. Эта устойчивость — результат нарушения активности каспазы-3 и цистеинил протеазы, регулирующих внутриклеточный биохимический путь, ответственный за апоптоз.
У некоторых пациентов вырабатываются белки внеклеточного матрикса и вещества, активирующие фибробласты, что приводит к фиброзу. В воспаленном органе саркоидозные гранулемы остаются на разных стадиях развития, регрессии и фиброза. Вопрос, почему у некоторых больных воспаление развивается, а у других регрессирует, пока остается без ответа.
Механизмы фиброза — следствие нарушения баланса между металлопротеиназами MMP-8 и MMP-9 и тканевыми ингибиторами металлопротеиназ — TIMP-1, а также повышенной активности альвеолярных макрофагов, продуцирующих повышенное количество фибронектина и лиганда хемокина CC18 – CCL18, стимулирующего выработку фибробластов легочными фибробластами. В свою очередь, коллаген в результате положительной обратной связи стимулирует макрофаги к выработке CCL18.
Риск интерстициального фиброза подтверждается повышенным процентом эозинофилов и нейтрофилов, которые в результате окислительного стресса, протеолиза и высвобождения токсичных белков способствуют неблагоприятным изменениям в легких. Признаки фиброза встречаются примерно у четверти пациентов.

Студент медицинского факультета УЛГУ. Интересы: современные медицинские технологии, открытия в области медицины, перспективы развития медицины в России и за рубежом.
- Запись опубликована: 28.10.2020
- Reading time: 4 минут чтения
Иммунная система человека существует для борьбы с болезнями. Но заболевание может начаться из-за ее сверхчувствительности. Одна из таких патологий – пурпура Геноха-Шенлейна. Это воспаление мелких кровеносных сосудов, которое сначала проявляется в виде сыпи.
Болезнь встречается относительно редко и в основном поражает детей в возрасте от двух до десяти лет. Еще реже встречается у взрослых. Пурпура Геноха-Шенлейна – аутоиммунное заболевание, поэтому болезнь не заразна. Однако может давать осложнения – боли, отеки суставов, кровотечение.
Что такое пурпура Геноха-Шенлейна?
Странное название Пурпура Шенлейн-Генох происходит от фамилий немецкого врача Иоганна Лукаса Шенлейна (1793-1864) и его ученика Эдуарда Генриха Геноха (1820-1910), которые описали болезнь.
Пурпура Шейлейна является васкулитом – болезнью, представляющей собой воспаление кровеносных сосудов, вызванное чрезмерной реакцией иммунной системы. Антитела иммуноглобулинов A (IgA), которые фактически должны бороться с патогенами, откладываются на стенках сосудов . Это вызывает воспалительную реакцию, которая делает мелкие кровеносные сосуды проницаемыми. На коже появляются точечные кровоизлияния, развивается типичная сыпь (пурпура).

Сосуд, пораженный васкулитом
Помимо кровеносных сосудов кожи, поражаются также сосуды суставов, желудочно-кишечного тракта и почек. Это приводит к боли, отекам, а иногда и к осложнениям. Самое опасное осложнение пурпуры – нефрит Геноха-Шенлейна. Это воспаления почек, которое может в долгосрочной перспективе привести к почечной недостаточности.
Болезнь обычно протекает спонтанно и проходит в течение трех – 16 недель. В отдельных случаях выздоровление может занять до года. Встречаются также случаи хронического течения пурпуры Геноха-Шенлейна. Заболевание часто протекает тяжелее у взрослых, чем у детей.
Особая проблема с этим заболеванием заключается в следующем: поскольку почки могут быть повреждены впоследствии, рекомендуется после постановки диагноза последующее наблюдение в течение двух лет.
Что вызывает пурпуру Геноха-Шенлейна?
Что именно вызывает пурпуру Геноха-Шенлейна, не совсем понятно. Но отмечено, что патологии часто предшествует инфекция верхних дыхательных путей. Другие причины – укусы насекомых и лекарства, особенно антибиотики и противовоспалительные препараты. Кроме того, как возможные триггеры обсуждаются варианты вакцинации и генетическая предрасположенность.
Симптомы пурпуры Геноха-Шенлейна
Заболевание начинается внезапно и прогрессирует поэтапно. Поскольку пурпура Геноха-Шенлейна поражает в основном мелкие кровеносные сосуды кожи, суставов, желудочно-кишечного тракта и почек, обычно возникают следующие симптомы:
- Кожа . Первый и наиболее очевидный симптом пурпуры – сыпь. Кровотечение на коже изначально выглядит как маленькие точки размером с булавочную головку, которые обычно распространяются по большой площади. Бугорки можно почувствовать при прикосновении, и их нельзя сдвинуть. Такие высыпания называются петехиями Henoch-Schönlein purpura petechiae и возникают в основном на ногах, поднимаясь от голеней к ягодицам, обычно симметрично.
- Суставы . В большинстве случаев жалобы возникают также в голеностопных и коленных суставах, иногда также в локтях. Суставы болят, опухают, иногда сильно чешутся. В некоторых случаях возникает воспаление суставов, обычно проходящее без последствий.
- Желудочно-кишечный тракт . Симптомы очень часто включают спастическую боль в животе, иногда тошноту и рвоту, а иногда и кровотечение из желудочно-кишечного тракта. Одно из возможных осложнений – так называемая инвагинация кишечника.
- Почки : через одну-две недели может появиться видимое или невидимое поражение почек. На это могут указывать выделение крови и белка с мочой, повышение артериального давления, головные боли, задержка воды и нарушение функции почек. Это может привести к серьезным осложнениям: тяжелому поражению почек, воспалению почек (нефрит Геноха-Шенлейна) и даже почечной недостаточности. Это осложнение чаще встречается у взрослых, чем у детей.
Другими симптомами могут быть: небольшое повышение температуры тела, головная боль, потеря аппетита.
Другие органы поражаются пурпурой Геноха-Шенлейна крайне редко:
- Мозг . Сосуды головного мозга поражаются очень редко. Патология приводит к головным болям, судорогам, параличу или нарушению сознания.
- Яички . Воспаление яичек, протекающее с болью и опухолью яичек, также встречается редко. Обратите внимание на осложнение – перекрут яичка (поворот яичка и семенного канатика).
Больные часто испытывают сильный зуд.
Частота проявления симптомов у больных:
- кожа – 100% случаев;
- желудочно-кишечный тракт – 80%;
- суставы – 75%;
- почки – около 50%.
Диагностика заболевания
Врач распознает пурпуру Геноха-Шенлейна на основании симптомов (клинический диагноз). Он исследует сыпь, определяя качество петехий. Диагноз может быть быстро поставлен, если есть сочетание с другими симптомами.
Чтобы определить степень поражения желудочно-кишечного тракта и почек или наличие осложнений, в лаборатории исследуются образцы крови, мочи и кала. Также проводится ультразвуковое исследование органов брюшной полости . Магнитно-резонансная томография необходима только в крайних случаях, при возможном поражении сосудов головного мозга.

Ультразвуковое исследование органов брюшной полости
Для определения степени заболевания могут потребоваться другие меры: лабораторные анализы, сонография, рентген, гастроскопия и колоноскопия.
В лаборатории при пурпуре определяют следующее:
- Кровь. Показатели воспаления несколько увеличены. Необходимо определить факторы свертывания крови и исключить другие сосудистые заболевания путем анализа на специфические антитела. Выявляется повышенный IgA, и могут быть обнаружены конгломераты антител (иммунные комплексы). О функции почек дает информацию значение креатинина.
- Моча . Повышенное количество красных кровяных телец и белка в моче может указывать на поражение почек.
- Стул . Если слизистая стенка кишечника повреждена, в стуле может быть скрытая кровь.
У взрослых отличить пурпуру Геноха-Шенлейна от других воспалительных заболеваний сосудов всегда сложнее. При сомнениях в диагнозе проводится биопсия и исследуются образцы тканей. При исследовании образца ткани покрасневшей кожи под микроскопом можно увидеть характерные изменения.
Как лечится пурпура Геноха-Шенлейна?
В большинстве случаев, особенно у детей, пурпура Геноха-Шенлейна не требует лечения. При лечении сложных случаев основное внимание уделяется облегчению симптомов.
Маленьким пациентам дают обезболивающие, например, парацетамол. При болях и зуде в суставах помогают болеутоляющие и противозудные мази .

Противозудная мазь
Во время острой фазы болезни следует избегать физических нагрузок. В то же время необходимо контролировать функцию почек.
В более тяжелых случаях с поражением пищеварительного тракта и / или почек пациенту назначают терапию кортизоном. Стационарное пребывание может потребоваться детям до двух лет, больным со сложным или более тяжелым течением.
Если заболевание является тяжелым или хроническим, чтобы подавить чрезмерную реакцию иммунной системы, можно вводить иммунодепрессанты. Также было замечено, что положительно сказывается на процессе заживления прием капсул с рыбьим жиром.
Как предотвратить пурпуру Геноха-Шенлейна?
Мер, предотвращающих или позволяющих избежать рецидива, пока не существует. У взрослых болезнь обостряется снова и снова. В этих случаях проводится профилактическое лечение иммунодепрессантами. Поддерживающая терапия может проводиться довольно долго.
Для наблюдения за пурпурой Геноха-Шенлейна важно регулярно проверять функцию почек в течение двух лет после постановки диагноза, особенно после нефрита Геноха-Шенлейна.
Шансы на выздоровление от пурпуры Геноха-Шенлейна
Заболевание обычно проходит самостоятельно без последствий. Прогноз обычно хороший. Однако рецидивы могут происходить в течение нескольких лет, что чаще встречается у взрослых, чем у детей. Чтобы вовремя обнаружить осложнение, после пурпуры Геноха-Шенлейна необходимо регулярно сдавать анализы мочи.
Если почки серьезно поражены, возникает почечная недостаточность. Таким пациентам, возможно, придется пройти диализ или даже пересадку почки.
Читайте также: