
Эпидермальная дисплазия у человека что это
Обновлено: 23.04.2024

У некоторых людей вследствие генетических мутаций страдает эктодерма — зародышевый листок (слой клеток), из которого на третьей неделе развития эмбриона возникают важные органы: кожа, центральная нервная система, хрусталик глаза, волосы, зубная эмаль, сальные и потовые железы, железы внутренней секреции, органы чувств. Патология получила название ангидротическая (гипогидротическая) эктодермальная дисплазия. Она проявляется отсутствием или аномальной формой зубов, бледностью и сухостью кожных покровов, редкими бровями, ресницами и волосами, отсутствием молочных желез у женщин и так далее. На данный момент специфическое лечение заболевания отсутствует.
Причины дисплазии
Заболевание достаточно редкое - 1 случай на 5000-10000 тысяч новорожденных. Оно регистрируется в разных странах мира у представителей различных рас, возникает из-за генных мутаций и может быть трех типов:
Х-сцепленный тип (синдром Криста-Сименса-Турена)
Самая распространенная форма патологии. От нее страдают не только мужчины, но и в меньшей степени - женщины-носительницы. Доказано, что ее провоцирует мутация гена EDA, расположенного на Х-хромосоме, вследствие которой нарушается структура белка эктодисплазина-а и нормальное развитие производных эктодермы.
Внимание! Как работает этот белок и почему ген мутирует, наверняка пока неизвестно.
Аутомсомно-рецессивный тип
Появляется вследствие мутации гена EDAR, расположенного на 2-й хромосоме и отвечающего за определения местоположения, размера и формы эктодермальных придатков (волосяных фолликул, зубов, потовых желез).
Внимание! При аутомсомно-рецессивном наследовании патологии человек заболевает, если получает проблемный ген от обоих родителей.
Аутосомно-доминантный тип
Редкая форма заболевания, причина которого – мутация гена TDARADD.
Внимание! При аутомсомно-доминантном наследовании патологии человек заболевает, если получает проблемный ген только от одного родителя, но он доминирует над здоровым. И такое происходит из поколения в поколение.
Симптомы дисплазии
Заболеванию чаще подвержены мальчики, что обусловлено сцепленным с полом наследованием (мальчики болеют чаще девочек). Девочки являются носительницами, болеют легче или вообще бессимптомно.
Обычно первые признаки патологии заметны еще с младенчества. Но у некоторых детей они носят невыраженный характер, а усугубляются лишь с годами (явно их может быть видно только к 2-3 годам).
Понять, что у ребенка эктодермальная дисплазия, можно при совокупности таких симптомов, как:
плохое потоотделение или его отсутствие (из-за проблем с потовыми железами);
относительно большая голова на фоне плохого роста;
отсутствие волос или их аномально медленный рост;
редкость ресниц и бровей;
неправильная форма ушных раковин;
неправильная форма лица вследствие патологий развития хрящей: большой лоб с выраженными бровями, запавшей переносицей;
запоздалое (от года до трех лет) прорезывание зубов, их аномальная форма (заостренная конусовидная) или отсутствие (с сохранением только клыков);
чрезмерная сухость кожи, постоянные экземы, бактериальные и грибковые кожные заболевания;
аномальная морщинистость лица;
отсутствие или недоразвитость грудных желез, сосков;
постоянные конъюнктивиты из-за плохой работы слезных желез.
Важно! Что касается интеллектуального развития ребенка, оно может отставать (диагностируется олигофрения) или находиться в норме.
При множественных зубных нарушениях в сочетании с расстройствами прочих анатомических структур эктодермального происхождения ребенок может признаваться инвалидом детства. Если же патологические изменения небольшие, не осложнены и протекают легко, в инвалидности откажут. Трудоспособность пациента оценивается не раньше, чем через год после определения диагноза, проведения необходимых лечебных и реабилитационных мероприятий.
Диагностика дисплазии
Врач (чаще всего – генетик) диагностирует наличие ангидротической эктодермальной дисплазии после визуального осмотра и генетических исследований:
прямого секвенирования последовательности гена EDA на предмет мутаций (позволяет определить генетические повреждений в ДНК, провоцирующие болезнь);
мультиплексной лигазной реакции (позволяет определить относительное количество копий определенных участков ДНК).
Немаловажным диагностическим этапом является анализ наследственного анамнеза. Особое внимание уделяется тому, какой объективный статус матери - были ли у нее проявления заболевания, как выражались.
Пациенту с дисплазией могут назначаться инструментальные исследования, например:
кожная биопсия (дает представление о состоянии потовыделительных желез);
микроскопия волосяной структуры;
рентген челюстей для анализа зубных зачатков.
Лечение дисплазии
Вылечит болезнь нельзя. Врачебное вмешательство сводится к поддержке ремиссии и нормальной жизнедеятельности пациента:
Чтобы человек не страдал от сухости кожных покровов, ему назначают лечебную и уходовую увлажняющую косметику, эмоленты. Возможно наружное применение кортикостероидных мазей. Также пациент занимается профилактикой вторичных бактериальных и грибковых инфекций. Некоторым прописывают фотолечение, но все индивидуально и зависит от состояния организма.
Проблемы с потоотделением и перегревом компенсируют влажными обтираниями и нахождением в прохладном помещении и обильным питьем в жару. Кроме того, постоянно отслеживают температуру тела.
Отсутствие или аномалии зубов исправляют у стоматолога-ортодонта: ставят корректирующие системы (брекеты, пластины), проводят синуслифтинг с дальнейшей имплантацией, зубное протезирование . Важно соблюдать меры профилактики кариеса, проводить раннюю реминерализирующую терапию и фторирование.
Проблемы со зрением решаются у офтальмолога. Если не намечается катаракты или других серьезных заболеваний, человеку прописывают увлажняющие капли. Если выявлена более серьезная патология – исправляют медикаментозно или хирургически.
Если эктодермальная дисплазия отягощена иммунодефицитом, пациент принимает препараты для поддержки иммунной системы, оберегает себя от инфекционных заболеваний, а в более сложных случаях участвует в пересадке гемопоэтических стволовых клеток.
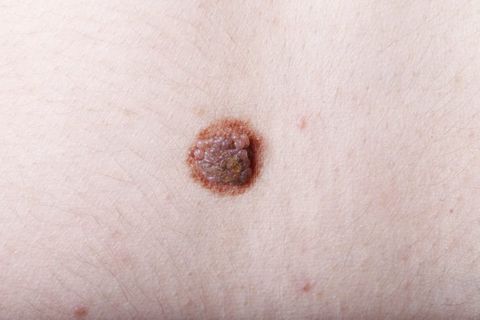
Невус (родинка, родимое пятно) – это доброкачественное опухолевидное новообразование, состоящее из меланоцитов (пигментных клеток). Он имеет яркую окраску – коричневую, черную, красную или фиолетовую и может быть плоским или возвышаться над кожей. Невусы бывают врожденными и приобретенными. И те, и другие наблюдаются у более чем 90% людей. В среднем у человека около 20 невусов на теле, однако это количество может колебаться от 3 до 100.Болезнь невус или нет? Сами по себе родинки не опасны и не причиняют никакого вреда здоровью. Их следует удалять лишь в том случае, если они доставляют эстетические или физические неудобства или расположены в местах, где их легко повредить механическим воздействием (например, трением об одежду). Однако невусы способны перерождаться в меланому (рак кожи) – злокачественное новообразование. Поэтому при наличии «подозрительных» родинок рекомендуется регулярно наблюдаться у дерматолога, а при признаках их перерождения в меланому незамедлительно обращаться к врачу.
Виды невусов и причины появления
Невус – узелок с обычно гладкой поверхностью. Некоторые родинки имеют ороговевшую или бородавчатую поверхность. Внутри таких образований могут расти волоски.

Невусы образуются из пигментных клеток, находящихся между двумя слоями кожи: эпидермисом и дермой. Меланоциты вырабатывают пигмент (меланин) под воздействием ультрафиолетовых лучей. Этим объясняется возникновение загара после длительного нахождения на солнце. Невусы появляются при размножении меланоцитов в слоях кожи.
Родинки могут быть:
- Врожденными – закладываются еще во внутриутробный период.
- Приобретенными – возникают под воздействием провоцирующих факторов.
Причины появления родинок во внутриутробный период:
- Наследственную предрасположенность «склонность» к образованию родинок способна передаваться генетически.
- Нарушения нормального течения беременности – токсикоз, угрозу прерывания.
- Аллергические реакции матери.
- Токсическое воздействие.
- Наличие у беременной патологий мочевыделительной системы.
- Лучевое облучение матери.
- Злоупотребление женщиной спиртными напитками (не только во время, но и до беременности).
- Прием гормональных препаратов до зачатия.
К провоцирующим факторам, повышающим вероятность появления приобретенных родинок, относятся:
- Ультрафиолетовое излучение.
- Гормональные нарушения.
- Травмы участка кожи.
- Вирусные и бактериальные инфекции.
- Воздействие радиации или рентгеновских лучей.
Справка! Так как возникновению невусов способствует изменение гормонального фона, новые родинки часто появляются в период полового созревания, во время беременности и менопаузы.
Существует теория, согласно которой все невусы являются врожденными, однако при появлении ребенка на свет они невидимы и проявляются позднее, в течение жизни. Действительно, всего 4-10% детей рождаются с невусами. В 90% случаев это мелкие родинки (менее 4 мм). 8% невусов у новорожденных – средних размеров, а с гигантскими появляются на свет всего 2% детей. Активно появляться они начинают после 5 лет жизни. К 15-16 годам у большинства подростков есть невусы. С возрастом количество родинок уменьшается. После 80 лет примерно у половины людей невусы на теле исчезают совсем.
Родинки классифицируются по размеру (диаметру):
- Мелкие – до 1,5 мм.
- Средние – до 10 мм.
- Крупные – более 10 мм.
- Гигантские – полностью охватывают какую-то часть тела.
Склонность к перерождению в меланому имеют, как правило, средние и крупные родинки.
Также врожденные невусы подразделяются на виды в зависимости от расположения:
- Эпидермальные – меланоциты скапливаются в верхнем слое кожи, эпидермисе.
- Внутридермальные – находятся в глубоких слоях кожи.
- Пограничные – расположены между эпидермисом и дермой.
В меланому способны перерождаться не все невусы кожи. Поэтому их делят на:
К меланомоопасным относят:
- Пограничный пигментный невус – плоский узелок с гладкой сухой поверхностью, без наличия в нем волосков. Чаще всего достигает 1 см в диаметре, однако его диаметр может колебаться от нескольких миллиметров до нескольких сантиметров. Пограничные невусы имеют разную окраску – от светло-коричневого до черного.
- Невус Ота – одно крупное пигментное пятно (или множественные, сливающиеся воедино) сине-черного цвета, обычно располагается на лице.
- Гигантский пигментный невус – относится к врожденным, растет вместе с ребенком и способен достигать в диаметре 10-40 см, имеет серую или черную окраску и неровную поверхность (может быть покрыт трещинами), в нем часто наблюдаются волоски.
- Невус Дюбрея – плоский, 2-6 см в диаметре, имеет неровные края и неравномерную серо-коричнево-синюю окраску и визуально напоминает географическую карту, поверхность может быть покрыта узелками и бляшками.
- Синий невус – новообразование с четкими границами, имеющее вид полусферы. Его диаметр редко превышает 1 см. Имеет голубую или синюю, иногда коричневую окраску. Поверхность мягкая, лишена волос.
- Диспластический невус – имеет размытые неровные границы, неравномерную окраску (смесь светло- и темно-коричневых оттенков), поверхность покрыта пятнами или узелками, в диаметре обычно превышает 6 мм.
Справка! Прилагательное «меланомоопасный» не означает, что доброкачественное образование обязательно станет злокачественным. Это говорит лишь о том, что у вышеперечисленных родинок есть такая способность. Если у вас есть меланомоопасный невус на лице или теле, за ним необходимо пристально наблюдать и обращаться к врачу при малейших подозрениях на начало перерождения.
Меланомонеопасными являются такие невусы, как:
- Внутридермальный меланоцитарный – наиболее распространенный вид родинки, представляет собой небольшое мягкое округлое коричневое образование, способен возникнуть на любом участке тела.
- Папилломатозный – напоминает папиллому, имеет несимметричную форму, чуть возвышается над поверхностью кожного покрова, может менять размеры в течение жизни (увеличиваться или уменьшаться), располагается на теле, лице или волосистой части головы.
- Фиброэпителиальный – мягкое новообразование на ножке, достигает до 15 мм в диаметре, имеет телесную, коричневую, черную, розовую или фиолетовую окраску, внутри родинки растут волоски.
- Монгольское пятно – врожденное плоское образование, имеющее синеватую окраску, в диаметре может достигать нескольких сантиметров (иногда десятков сантиметров), чаще всего располагается на ягодицах или в области крестца, обычно проходит само собой к 5-13 годам.
Признаки перерождения невуса в меланому
Симптомами озлокачествления невуса являются:
- Изменение цвета (снижение или повышение пигментации).
- Уплотнение родинки.
- Разрастание новообразования – в диаметре или в высоту.
- Покраснение.
- Наличие на родинке язв.
- Кровотечения из невуса.
- Возникновение вокруг родинки или родимого пятна черных точек.
- Выпадение из невуса волос.
- Ощущение тепла, зуда или жжения в области новообразования.
- Увеличение регионарных лимфатических узлов.
При наличии таких признаков следует незамедлительно обратиться к дерматологу и онкологу.
Причины перерождения невуса в меланому
Спровоцировать озлокачествление новообразования могут:
- Травмы (ушибы, порезы и пр.).
- Некачественное и неполное удаление невуса.
- Воздействие ультрафиолетового или радиоактивного излучения.
Диагностика

В комплекс диагностических мероприятий входят:
- Осмотр дерматолога/онколога.
- Дерматоскопия – микроскопическое исследование.
- УЗИ новообразования – для оценки глубины прорастания невуса в слои кожи.
Справка! Биопсия «живой» родинки не осуществляется, так как вследствие этой процедуры невус травмируется, а механическое повреждение способно спровоцировать перерождение в меланому. Поэтому гистологическое исследование проводят после удаления новообразования.
Лечение
Лечение невусов в основном хирургическое. Удалить новообразование можно с помощью:
- Обычного хирургического вмешательства.
- Лазера.
- Электрокоагуляции.
- Радионожа.
- Криодеструкции.
Метод устранения потенциально опасного невуса выбирается врачом в зависимости от характера новообразования, его величины, расположения, состояния организма пациента.
Ангидротическая эктодермальная дисплазия – это наследственное заболевание, проявляющееся генетическим нарушением развития производных эктодермы (кожи, желез внешней секреции, волос, зубов). Симптомами патологии являются аномалии развития или отсутствие зубов, сухая тонкая кожа, выраженная гипоплазия потовых и сальных желез, редкость волос или алопеция, иногда агенезия молочных желез. Диагностика производится врачом-генетиком на основании характерных внешних проявлений заболевания, а также генетических исследований и изучения наследственного анамнеза. Специфического лечения ангидротической эктодермальной дисплазии не существует.
МКБ-10

Общие сведения
Ангидротическая эктодермальная дисплазия (синдром Криста-Сименса-Турена, синдром Weech) является наследственным заболеванием, при котором происходит генетически обусловленное нарушение развития наружного зародышевого листка (эктодермы). В результате этого основные симптомы патологии затрагивают производные эктодермы – кожу, волосы, зубы, некоторые хрящи, потовые, сальные и молочные железы. Характерный для ангидротической эктодермальной дисплазии симптомомкомплекс независимо друг от друга описывали Дж. Турен в 1848 году, стоматолог Дж. Крист в 1913-м и дерматовенеролог Х. Сименс в 1929-м году.
Первоначально считалось, что наследование заболевания происходит исключительно по сцепленному с Х-хромосомой механизму, в настоящее время выявлены как аутосомно-рецессивные, так и аутосомно-доминантные формы дисплазии. Встречаемость заболевания – 1 случай на 5000-10000 тысяч новорожденных, с учетом того, что статистически чаще встречается сцепленное с полом наследование, половое распределение среди больных сильно смещено в сторону мужского пола.
Причины
Этиология ангидротической эктодермальной дисплазии заключается в наличии мутаций определенных генов. Различают формы синдрома Криста-Сименса-Турена с Х-сцепленным, аутосомно-доминантным и аутосомно-рецессивным наследованием:
- Х-сцепленный тип. Причиной наиболее распространенной формы заболевания является повреждение гена EDA, расположенного на Х-хромосоме. Он кодирует белок под названием эктодисплазин-а, нарушения в структуре которого и приводят к патологическому развитию производных эктодермы. В настоящий момент, как функции этого белка, так и патогенез нарушений при мутации гена EDA неизвестны. Симптомы выявляются не только у мужчин, но и у женщин-носительниц, в основном, в более легкой степени. У таких женщин наблюдается сухость кожи, более ранее развитие морщин, тонкие сухие волосы, деформации и патологии зубов. Также нередко возникают проблемы с грудным вскармливанием ребенка. Все это позволяет говорить о том, что некоторые мутации гена EDA обладают свойствами неполного доминирования.
- Аутомсомно-рецессивный тип. Кроме того, к характерному симптомокомплексу синдрома Криста-Сименса-Турена приводят мутации в гене EDAR, кодирующем один из рецепторов к фактору некроза опухоли. Данный ген расположен на 2-й хромосоме и наследуется по аутосомно-рецессивному типу. Как и в предыдущем случае, патогенез при этой форме заболевания не изучен.
- Аутосомно-доминантный тип. Редкая форма ангидротической эктодермальной дисплазии, передающаяся по аутосомно-доминантному механизму. Ее причиной служат мутации гена TDARADD, который кодирует белок рецептор к экзодисплазину-а и расположен на 1-й хромосоме. По всей видимости, патогенез нарушений в этом случае аналогичен таковому при распространенной сцепленной с полом форме синдрома.
Симптомы
Возникшая гипоплазия кожи и многих типов желез (потовых, слезных, молочных) ведет к каскаду разнообразных нарушений. Практически полное отсутствие потовых желез становится причиной легкого развития гипертермии, что особенно опасно в детском возрасте – именно из-за последствий перегрева в раннем детстве умирает почти треть больных ангидротической эктодермальной дисплазией. В результате пониженной активности слезных желез достаточно часто возникают конъюнктивиты, которые осложняются кератитом и катарактой.
Гипопластическая кожа довольно часто подвержена экземе, вторичным бактериальным и грибковым инфекциям. Нарушения развития эктодермы отражаются и на хрящевых и костных элементах – увеличивается размер лобной кости, на ней формируются заметные надбровные дуги. Переносица запавшая, крылья носа, как правило, недоразвиты, деформируются и ушные раковины.
Типичны аномалии зубов, которые приобретают коническую форму, часто бывают недоразвиты, возможно отсутствие одного или целой группы зубов; характерным признаком ангидротической эктодермальной дисплазии при этом является сохранение клыков. Из-за отсутствия или аномального положения зубов нередко развиваются дефекты речи.
Интеллектуальное развитие ребенка может отставать от возрастной нормы, но в ряде случаев умственные способности взрослого с данным синдромом не уступают таковым у здорового человека. Часто наблюдается отсутствие молочных желез и сосков (либо их аномальные форма и расположение). Иногда ангидротическая эктодермальная дисплазия осложняется врожденной глухотой.
Диагностика
Диагностика заболевания производится на основе обследования ребенка у генетика, ДНК-исследования и изучения наследственного анамнеза. При осмотре пациента на патологию указывают характерные сочетания признаков и объективный статус больного. Генетическое определение заболевания сводится к прямому секвенированию последовательности гена EDA с целью выявления мутаций; изучение других, более редких мутаций, ассоциированных с ангидротической эктодермальной дисплазией, в настоящий момент не производится.
При изучении наследственного анамнеза особое внимание уделяют объективному статусу матери – нередко у нее, как у носительницы мутантного гена, обнаруживаются стигмы дизэмбриогенеза. К ним относят сухость кожи, ослабленные тонкие волосы, гипоплазия молочных желез, из-за чего возникают проблемы с кормлением ребенка.
Генетическая диагностика носительства мутантной формы гена EDA сопряжена с определенными сложностями, так как метод прямого секвенирования в таком случае часто дает ложноотрицательные результаты. Поэтому для этой цели используются другие методики генетического анализа – например, мультиплексная лигазная реакция.
Лечение ангидротической эктодермальной дисплазии
Специфического лечения данной патологии не существует, терапия сводится к поддержке нормальной жизнедеятельности и профилактике осложнений. Для увлажнения кожи используют специальные фармацевтические или косметологические кремы, аномалии зубов исправляют при помощи протезирования. Из-за нарушения потоотделения крайне опасным становится перегрев, поэтому особую осторожность необходимо проявлять в летние жаркие месяцы. Больных ангидротической эктодермальной дисплазией в этот период желательно содержать в кондиционированном помещении, можно оборачивать влажной простыней для увлажнения, давать обильное питье. Также проводят лечение и профилактику вторичных бактериальных и грибковых инфекций кожи, иммуномодулирующую терапию. Для профилактики глазных нарушений необходимо регулярное использование увлажняющих капель.
Прогноз
Прогноз заболевания зависит от степени выраженности сопутствующих нарушений и своевременного выявления патологии. В большинстве случаев, если диагноз был установлен в раннем возрасте ребенка и были предприняты профилактические меры (борьба с перегревом, вторичными инфекциями), то прогноз в целом благоприятный. В плане интеллектуального развития прогноз чаще всего неоднозначный – с одинаковой вероятностью возможна как умственная отсталость (олигофрения), так и сохранение когнитивного развития.
1. Ангидротическая эктодермальная дисплазия/ Иванова И.Н., Сердюкова Е.А., Иконникова Т.И.// Российский журнал кожных и венерических болезней. - 2012.
2. Ангидротическая эктодермальная дисплазия у новорожденного ребенка (клиническое наблюдение)/ Ахмина Н.И. и др.// РМЖ. Мать и дитя. - 2020.
3. Синдром Криста—Сименса—Турена/ Васина Т.Н. и др.// Российский вестник перинатологии и педиатрии. - 2013.

Весь контент iLive проверяется медицинскими экспертами, чтобы обеспечить максимально возможную точность и соответствие фактам.
У нас есть строгие правила по выбору источников информации и мы ссылаемся только на авторитетные сайты, академические исследовательские институты и, по возможности, доказанные медицинские исследования. Обратите внимание, что цифры в скобках ([1], [2] и т. д.) являются интерактивными ссылками на такие исследования.
Если вы считаете, что какой-либо из наших материалов является неточным, устаревшим или иным образом сомнительным, выберите его и нажмите Ctrl + Enter.
Дисплазии эктодермальные - группа наследственных заболеваний, обусловленных ненормальным развитием эктодермы, и сочетающаяся с различными изменениями эпидермиса и придатков кожи.
Причины и патогенез дисплазии эктодермальной заболевания окончательно не установлены. В настоящее время в литературе описано несколько десятков дерматозов эктодермального происхождения. Принято считать, что следующие две формы являются классическими: ангидротическая и гидротическая. Ангидротическая форма часто наследуется Х-сцепленно рецессивно, реже - по аутосомно-рецессивному типу. Описаны семейные случаи заболевания. Гидротическая форма эктодермальной дисплазии передается по наследству аутосомно-доминантно. Некоторые ученые считают, что под влиянием рентгеновых лучей, вирусных инфекций, химических соединений может произойти нарушение дисульфидных связей во фракции L-кератинового матрикса.
Симптомы дисплазии эктодермальной. Обе формы эктодермальной дисплазии развиваются вскоре после рождения или в раннем детском возрасте с формированием полной клинической картины к периоду полового созревания.
Ангидротическая эктодермальная дисплазия (синдром Криста-Сименса) - гетерогенное заболевание, в большинстве случаев наследуемое рецессивно, сцеплено с Х-хромосомой, локализация гена - Xql2-ql3. Полная клиническая картина, включающая такие основные признаки, как ангидроз, гипотрихоз и гиподонтия, наблюдается, за редким исключением, только у мужчин.
Ангидротическая форма эктодермальной дисплазии встречается преимущественно у лиц мужского пола. Характерными признаками является следующая триада: гипо- или ангидроз, гипотрихоз, гиподонтия. Вначале у больных отмечается нарушение терморегуляции за счет гипо- или аплазии потовых желез. Больные ангидротической формой страдают гипертермией, что может служить тяжелыми последствиями при нераспознанных случаях. Наблюдается снижение или отсутствие потоотделения по всему кожному покрову, за исключением мест локализации апокриновых желез, где оно в большей или меньшей степени может сохраняться. При осмотре отмечаются выраженная сухость и истончение кожи. у некоторых больных - ихтиозиформпое шелушение, фолликулярный гиперкератоз, слабо заметная кератодермия ладоней и подошв. Из-за сухости развиваются конъюнктивиты, ларингиты, фарингиты, риниты, стоматиты. Ресницы, брови редкие, оволосение в лобковой области и подмышечных впадинах отсутствует. Пушковые волосы обычно не поражены. Характерен внешний вид больных: усталое, старческое выражение лица, низкий рост, большой квадратный череп с выступающими лобными буграми, массивный подбородок, надбровные дуги, высокие и широкие скуловые кости, впалые щеки, выпуклые губы, большие оттопыренные уши (уши сатира), седловидный нос, поредение волос вплоть до алопеции. Зубы прорезываются поздно, долго сохраняются в стадии молочных, имеется большое расстояние между верхними резцами, могут быть не в полном количестве или даже отсутствовать полностью, часто деформированы. Умственное развитие у большинства больных нормальное, иногда выявляются признаки снижения интеллекта, потеря слуха, подверженность инфекциям. У женщин заболевание протекает в смягченной форме в виде нерезко выраженных зубных аномалий, очаговых расстройств потоотделения, слабого развития молочных желез.
Гидротическая эктодермальная дисплазия (синдром Клоустона) наследуется аутосомно-доминантно, проявляется двумя основными признаками - гипотрихозом и аномалиями ногтей. Предполагается, что биохимический дефект заключается в уменьшении числа дисульфидных связей во фракции а-кератинового матрикса.
При эктодермальной гидротической дисплазии основными признаками являются дистрофия ногтей и гипотрихоз, в меньшей мере - ладонно-подошвенный кератоз, нарушение пигментации. Потоотделение, как правило, не нарушено. Изменение ногтей является основным, наиболее частым, а иногда и единственным признаком болезни. Ногтевые пластинки растут медленно, утолщены, хрупкие, легко обламываются, коричневого цвета, деформированы, местами атрофированы. По краю ногти как бы изъедены, расщеплены, продольно исчерчены. Может наступить онихолизис.
Волосы истончены, сухие, легко обламываются, поредевшие, иногда до степени тотальной алопеции. Могут наблюдаться перекрученные волосы, узловатая ломкость волос, их расщепление, выпадение бровей и ресниц, скудное оволосение в подмышечных впадинах и на лобке. Имеются очаги гипо- и депигментации, напоминающие иногда витилигинозные пятна. Обнаруживают утолщение концевых фаланг, изменение зубов обычно отсутствует либо незначительно. С возрастом явление ксратодермии усиливается.
Гистопатология. При ангидротической форме дисплазии отмечаются отсутствие потовых желез. нормальное строение волосяных фолликулов и сальных желез. При гидротической дисплазии наблюдается гипоплазия волос и сальных желез.
Патоморфология. При гистологическом исследовании определяются истончение эпидермиса, гиперкератоз с образованием роговых пробок в устьях волосяных фолликулов, гипоплазия волосяных фолликулов и сальных желез. При ангидротической форме потовые железы отсутствуют, при гидротической - в пределах нормы. Однако признаки потоотделения, появляющиеся в процессе лечения тигазоном больных ангидротической эктодермальной дисплазией, свидетельствуют о том, что количество потовых желез лишь уменьшено и/или они функционально неполноценны (собственные наблюдения). Изменения волос при обоих видах эктодермальной дисплазии сходны. Они скручены, изменены по типу узловатого трихорексиса. Часто стержни волос меньшего, чем в норме, диаметра, отмечаются асимметрия волосяных фолликулов, пролиферация клеток их наружного корневого влагалища. Среди пролиферирующих клеток встречаются дискератотические; отмечается новообразование волосяных фолликулов из клеток наружного корневого влагалища. Внутреннее корневое влагалище утолщено, деформировано. При сканирующей электронной микроскопии обнаружены уменьшение диаметра стержня волоса, изменение его формы oт круглой до уплотенной; чешуйки кутикулы атрофичны или отсутствуют. Слущивание клеток кутикулы более заметно по направлению к верхушке волоса. Мозговое вещество волоса имеет грубоволокнистое строение. Аномалии кератинизании, возможно, обусловлены генетическим изменением последовательности аминокислот в белках коркового и мозгового веществ волоса, что приводит к нарушению окисления тиоловых групп между полипептидами филаментов коркового вещества волоса.
Дифференциальный диагноз необходимо проводить с прогериями, врожденным сифилисом, врожденной пахионихией.
Лечение дисплазии эктодермальной симптоматическое. Необходимо предохранять больных с ангидротической формой от перегревания.

[1], [2]
Весь контент iLive проверяется медицинскими экспертами, чтобы обеспечить максимально возможную точность и соответствие фактам.
У нас есть строгие правила по выбору источников информации и мы ссылаемся только на авторитетные сайты, академические исследовательские институты и, по возможности, доказанные медицинские исследования. Обратите внимание, что цифры в скобках ([1], [2] и т. д.) являются интерактивными ссылками на такие исследования.
Если вы считаете, что какой-либо из наших материалов является неточным, устаревшим или иным образом сомнительным, выберите его и нажмите Ctrl + Enter.

О таком синдроме, как дисплазия соединительной ткани, говорят тогда, когда организм человека с рождения склонен к нарушениям формирования хрящевой ткани суставов, а также прочих тканей. Малыша, страдающего дисплазией, выявить относительно легко: он отличается необычной гибкостью, его суставы без проблем прогибаются в разные стороны.
У пациентов с дисплазией соединительной ткани уже в молодом возрасте развивается ранний остеохондроз, отмечаются нарушения зрения, пороки клапанов сердца. Соответственно, такие люди быстро обретают разные проблемы со здоровьем – в частности, с опорно-двигательным аппаратом.
Код по МКБ-10
Эпидемиология
О дисплазии соединительной ткани говорят в случаях, когда имеются признаки нарушенного соединительнотканного развития на эмбриональном и постнатальном этапе, и эти нарушения вызывают сбой гомеостаза. Расстройство происходит на уровне тканей, органов и всего организма в целом: отмечаются всевозможные морфофункциональные патологии.
Распространенность и частота заболеваний соединительной ткани, о которых сообщают, весьма различны, в зависимости от различий в методологии исследования. [1] Проблема недифференцированной дисплазии соединительной ткани (нДСТ) является актуальной по причине значительной частоты встречаемости данной патологии в популяции взрослого населения в целом, и в частности, среди женщин репродуктивного возраста (7-8%). [2] За помощью медиков пациенты с дисплазией обращаются в шесть раз чаще, нежели больные другими заболеваниями.
Заболеваемость не связана с половой и расовой принадлежностью пациентов.
Причины дисплазии соединительной ткани
Дисплазия соединительной ткани – это синдром, который включает в себя обширный ряд патологий. Причинами выступают расстройства, связанные с генетическими нарушениями построения коллагеновых соединительнотканных волокон. Процесс преимущественно захватывает костную ткань, связочный и сухожильный аппарат и кожные покровы.
Базовым механизмом соединительнотканных нарушений являются генные мутации. Особую роль играют изменения в генах, ответственных за продукцию основного белкового вещества, составляющего соединительную ткань – речь идет о коллагене (иногда – о фибриллине). Когда в ходе формирования белковых волокон происходят болезненные изменения, то они становятся менее прочными, неспособными выдержать нагрузку. Дополнительным фактором развития синдрома может стать недостача магния в организме.
Факторы риска
Учеными доказано, что развитию дисплазии соединительной ткани у ребенка способствуют такие факторы:
- анемия у матери при беременности;
- угроза прерывания беременности;
- хроническая недостача кислорода у плода;
- хроническая фетоплацентарная недостаточность;
- сильный или продолжительный токсикоз, гестоз;
- сопутствующие беременности хронические патологии (заболевания эндокринной системы, почек, органов желудочно-кишечного тракта или респираторных путей).
Патогенез
Гетерозиготные мутации в гене коллагена типа II (COL2A1) приводят к группе скелетных дисплазий, известных как коллагенопатия типа II (COL2pathy). [3], [4], [5] Цепи proα1 (I) и proα2 (I) коллагена 1 кодируются генами COL1A1 и COL1A2 соответственно; Количественные или качественные дефекты синтеза коллагена I типа обычно проявляются в виде коллагенопатии I типа и несовершенного остеогенеза. Большинство пациентов (около 90%) с клиническим диагнозом несовершенного остеогенеза имеют мутацию в генах COL1A1 или COL1A2 с аутосомно-доминантным типом наследования. Шесть других генов, CRTAP, LEPRE1, FKBP10, PP1B, SP7 / Osterix (OSX) и SERPINH1, связаны с аутосомно-рецессивными формами. [6], [7], [8]
Базовый механизм развития дисплазии соединительной ткани, равно как и недифференцированной формы заболевания, обусловлен генной мутацией, с вовлечением генов, отвечающих за выработку и диссимиляцию строительных белковых компонентов соединительной ткани, либо ферментных веществ, принимающих участие в указанных процессах. Изменяется количественное формирование качественных составляющих экстрацеллюлярного матрикса, расстраивается фибриллогенез. Генетические детерминанты осуществляются в зависимости от внешних факторов, или практически не зависят от них: это отмечается при дисплазии и недифференцированной дисплазии соответственно. Для соединительнотканной дисплазии присуща полигенность и мультифакторность (патология с генетической предрасположенностью): речь идет о мутации сразу многих генов, а случайное перераспределение отцовских и материнских аллелей постоянно влечет за собой образование следующего единственного в своем роде генотипа.
Факторы при рождении – например, витаминная или макро и микроэлементарная недостача – становятся базовыми причинами, создающими предпосылки для развития дисплазии соединительной ткани. Витамины B-группы стабилизируют обмен белков, аскорбиновая кислота с токоферолом потенцируют адекватную выработку коллагена, а также выступают в роли антиоксидантов. Микро и макроэлементы – медь, бор, цинк и кремний, фтор и кальций, марганец и магний, ванадий, фосфор и селен – выступают кофакторами ферментных веществ, стимулирующих выработку коллагена и насыщение костей минералами. Немаловажно и их участие в электролитном обмене и поддержании кислотно-щелочного равновесия. Калиевые, магниевые и цинк-ионы поддерживают костный рост и усиливают минеральную концентрацию ткани кости. В развитии заболевания любой из указанных факторов имеет первостепенное значение. [9]
Симптомы дисплазии соединительной ткани
Первые признаки дисплазии соединительной ткани проявляются ещё в раннем детском возрасте. Это может быть, как чрезмерная гибкость и гиперподвижность, так и ограниченная мобильность суставов по типу контрактур. Случаются также физические дефекты развития (карликовость), связочная слабость, хрупкие костные ткани, различные искривления позвоночника, плоскостопие, деформированная грудная клетка и пр.
Признаки дисплазии отмечаются и по отношению к другим органам: болезнь может поражать сердце, сосудистую сеть, глаза.
Часто страдает позвоночный столб: позвонки смещаются настолько, что при малейшем движении происходит сдавливание сосудов, ущемляются нервные окончания, возникают боли, нарушается сознание. [10]
Клиническая картина заболевания поражает своим разнообразием, и в этом заключается огромный «минус», поскольку идентифицировать патологию становится очень сложно. Поэтому врачи вынуждены прибегать сразу к нескольким методам лабораторной диагностики, а также к инструментальным видам исследования.
Фенотипические признаки при дисплазии соединительной ткани не всегда присутствуют с рождения и могут проявляться на протяжении всего жизненного периода. Со временем, через годы, чаще всего – под воздействием определенных неблагоприятных условий численность диспластических симптомов и их выраженность увеличивается и усиливается, поскольку первичные нарушения гомеостаза нарастают. В данном случае неблагоприятными условиями могут стать неправильное питание, плохая экология, регулярные интеркуррентные патологии, частые стрессы и пр. Первоочередно затрагивается постоянство присутствия микро и макроэлементов, которые непосредственно участвуют в процессах коллагеновой выработки, а также в регуляции ферментной активности, необходимой для быстрого и качественного синтеза.
В общем, указанные процессы преимущественно зависимы от равновесия содержания кальция и магния в организме. Например, недостача магния на фоне нормы или превышения уровня кальция приводит к повышению активности протеолитических ферментных веществ, которые вызывают коллагеновую деградацию. Как следствие – тяжелая клиническая картина дисплазии соединительной ткани.
Магний регулирует утилизацию кальция в организме. При дефиците магния кальций откладывается в костных и мягких тканях разных органов. При избытке магния кальций начинает плохо усваиваться и выводиться из организма.
Длительный недостаток магния может вызывать признаки ангиоспазма, повышения артериального давления, миокардиальной дистрофии, тахикардии, аритмии, повышенного тромбообразования. Возможны психоневрологические расстройства: невнимательность, депрессия, фобии или тревожные состояния, вегетативные нарушения, головные боли и головокружения, бессонница, онемения конечностей. Висцеральные признаки могут обнаруживаться в виде бронхо или ларингоспазмов, спастических запоров или гиперкинетических поносов, диспепсии, дискинезий желчного пузыря, болей в животе.
Хроническая магниевая недостача дополнительно проявляется пониженным тонусом мускулатуры, малой плотностью костной ткани.
Морфометрическая характеристика черепа при дисплазии соединительной ткани может изменяться из-за особенностей гемостаза. У больных зачастую диагностируются аортальные аневризмы, сопровождающиеся развитием хронического диссеминированного внутрисосудистого свертывания крови, как результата застоя в аневризменной полости и создания турбулентного тока в аорте. Возможно формирование ишемических поражений мозга, субарахноидальные, паренхиматозные кровоизлияния.
На сегодняшний день специалисты определили ряд фенотипических признаков дисплазии СТ. Их условно можно поделить на визуальные (те, которые можно заметить внешне) и на такие, которые обнаруживаются только по результатам тщательного внутреннего обследования.
У большинства пациентов наблюдается:
- высокая утомляемость, частая беспричинная усталость;
- частые простудные заболевания, ОРВИ;
- склонность к кровоточивости (большие потери крови при удалении зубов, при травмах, во время менструации у женщин);
- головокружения и боли в голове.
Более чем у 30% пациентов наблюдается так называемое «готическое небо», нарушения прикуса, гиперподвижность суставов, преждевременное старение лица, плоскостопие.
Боли при дисплазии соединительной ткани беспокоят в зависимости от того, какой орган поражен более других. Так, часто могут беспокоить периодические и недлительные боли в сердце, за грудиной и в области подреберья, спастические боли по ходу кишечника, головная боль. Неприятные болезненные ощущения в суставах появляются на этапе присоединения остеохондроза. Если имеются деформации грудной клетки или позвоночного столба, то боли в спине и груди возникают при продолжительном стоянии, ходьбе, либо даже в сидячем положении.
Страдают ли зубы при дисплазии соединительной ткани? Было проведено немало исследований, поскольку ученые пытались связать изменение качества зубной эмали с дисплазией соединительной ткани, что позволило бы более точно устанавливать диагноз заболевания. По итогам таких работ были обнаружены нарушения минерализации и формирования эмали зубов у пациентов с признаками соединительнотканной дисплазии. Это обусловлено недостаточной плотностью упаковки эмалевых призм на единицу объема. Кроме этого, призмы хаотично расположены, а органический матрикс слабо организован и минерализован. Склонность к неправильному развитию зубов и вероятность связанных с этим патологий определяется индивидуально, поскольку проявляется не у всех больных данным заболеванием.
Читайте также: