
Что такое повышенная растяжимость кожи
Обновлено: 29.04.2024
Гиперэластическая кожа или синдром Элерса-Данлоса. Пигментная крапивница
Этот синдром характеризуется: 1) усиленной эластичностью кожи; 2) чрезмерной гибкостью суставов; 3) ломкостью кожи с образованием рубцов и 4) развитием изюмоподобных псевдоопухолей. Псевдоопухоли мягкие и пигментированные; поверхность их морщиниста.
Гистопатология синдрома Элерса-Данлоса. Дегенеративные изменения обнаруживаются как в коллагеновой, так и в эластической ткани. Многие авторы полагают, что изменения коллагена являются первичными и доминирующими [Кортинг и Готтрон (Коrting, Gottron)]. Коллагеновые пучки становятся атрофичными; они расщепляются и отделяются друг от друга отеком.
Эластические волокна в некоторых случаях бывают нормальными, но в большинстве случаев они разрываются и имеют вид комков. Количество эластической ткани часто является увеличенным; по-видимому, однако, это увеличение является не истинным, а относительным (результат атрофии коллагена). Количество капилляров увеличено, их просветы расширены. Могут наблюдаться большие кистозные пространства, представляющие собой лимфангиэктатические полости (Кортинг и Готтрон).
Изюмоподобные псевдоопухоли, которые часто представляют собой часть данного синдрома, развиваются на участках травматических геморрагии и состоят из скопления гигантских клеток инородных тел [Рончезе (Ronchese)] или из пролиферированной соединительной ткани с большим количеством сосудов в ней.
Плотные подкожные узлы содержат обызвествленный некротический жир или слизистую массу, внедренную в толстую фиброзную капсулу [Джонсон и Фоллс (Falls)].

Пигментная крапивница
Это заболевание характеризуется большим количеством коричневых пятен, которые могут располагаться на всех участках кожного покрова. При трении пятен тупым предметом они превращаются в волдыри. В редких случаях поражение состоит из мягких узелков и бляшек.
Гистопатология пигментной крапивницы. Гистологическая картина характеризуется инфильтратом, состоящим преимущественно из тучных клеток. При пятнистом типе высыпаний тучные клетки расположены диффузно в верхней трети дермы. Некоторые тучные клетки имеют круглую или овальную форму, большинство же веретенообразной формы. Тучные клетки имеют тенденцию располагаться вокруг капилляров подсосочкового слоя и вблизи придатков кожи.
С возрастом количество тучных клеток уменьшается, поэтому при гистопатологическом исследовании пигментной крапивницы у взрослых больных они могут отсутствовать.
При узелковом типе пигментной крапивницы тучные клетки расположены компактно в виде опухолеподобных скоплений в верхней части дермы. Инфильтрат может через всю дерму проникать в подкожную жировую клетчатку.
Если тучные клетки расположены в виде густых агрегатов, они чаще имеют кубическую, чем веретенообразную форму; протоплазма их обильная и слегка эозинофильная. Вследствие своей формы и обилия протоплазмы они не напоминают никакие иные клетки, и диагноз может быть установлен без применения специальных окрасок.
Если биопсия производилась вскоре после того, как элементы подвергались трению, в препарате обнаруживается отек, большое количество эозинофилов и сморщивание тучных клеток со значительным уменьшением количества зерен в них, что указывает на выхождение зерен из тучных клеток [Дреннэн (Drennan)]. Иногда наблюдается даже разрушение и временное исчезновение тучных клеток, что может объяснить некоторые описанные в литературе типичные случаи пигментной крапивницы без обнаружения тучных клеток [Дреннэн и Бир (Веаге)].
Информация на сайте подлежит консультации лечащим врачом и не заменяет очной консультации с ним.
См. подробнее в пользовательском соглашении.
Синдром гипермобильности суставов – это заболевание, при котором наблюдается избыточная подвижность, сочетающаяся с патологическими симптомами со стороны опорно-двигательного аппарата. Обычно пациентов беспокоят суставные и мышечные боли. Возможны синовиты, бурситы, энтезопатии, повторные подвывихи, ранние артрозы, другие проявления. Диагноз выставляется на основании анамнеза, данных клинического осмотра. Для исключения иных заболеваний назначаются лабораторные и инструментальные исследования. Лечение включает медикаментозную терапию, ЛФК, коррекцию режима дня и двигательной активности.
МКБ-10

Общие сведения
Синдром гипермобильности суставов (СГС) – патологическое состояние, которое следует отличать от асимптомной гипермобильности суставов (ГС), не сопровождающейся клиническими проявлениями. Распространенность СГС в популяции составляет около 4%. Женщины болеют чаще мужчин (5,6% и 1,9% соответственно). У 65% у пациентов имеются близкие родственники, страдающие тем же заболеванием, что свидетельствует о наследственной природе патологии. СГС ассоциируется с повышенным риском развития остеоартроза и других болезней костно-мышечной системы.
Причины
Генерализованная гипермобильность суставов – широко распространенное состояние. По данным исследователей, увеличение объема движений по сравнению со среднестатистической нормой выявляется у 6,9-31,5% жителей планеты, чаще – у женщин, уроженцев Азии, Африки, Ближнего Востока. В основе избыточной подвижности лежат мутации генов, отвечающих за формирование коллагена и тенаскина Х, которые входят в состав связок, сухожилий и суставных капсул.
Данная индивидуальная особенность передается по наследству, преимущественно по женской линии. Это нарушение – дисплазия соединительной ткани (ДСТ) потенцируется влиянием множества генов, при этом имеет значение уникальное сочетание аллелей обоих родителей, что объясняет значительную вариабельность проявлений ГС, неопределенную вероятность развития СГС, а также наличие или отсутствие симптомов соединительнотканной недостаточности со стороны других органов и систем.
Патогенез
Изменение структуры соединительной ткани приводит к снижению ее прочности и, как следствие – к повышению вероятности микротравм при физических нагрузках. Наряду с повторными микротравмами имеет значение снижение порога болевой чувствительности и наличие ортопедических аномалий, которые часто выявляются у больных с ДСТ – плоскостопия, дисплазии тазобедренных суставов, спондилолистеза, сколиоза.
Определенную роль играют вегетососудистые расстройства, которые также часто сопутствуют ДСТ. Все перечисленное обуславливает появление болей в мышцах и суставах на фоне физической активности. С возрастом проявления СГС усугубляются, потому что к врожденным аномалиям добавляются приобретенные патологические изменения – менископатии, энтезопатии, тендиниты, остеохондроз, развивающиеся на фоне базовых нарушений строения ОДС, повторных микротравм.
Симптомы
Первые симптомы обычно появляются в молодом возрасте. Больные предъявляют жалобы на мышечно-суставные боли после умеренной физической активности или незначительной травмы. Как правило, наибольшая интенсивность болей отмечается в голеностопных и коленных суставах. Признаки воспаления отсутствуют. Рецидивирующий характер боли вынуждает пациентов ограничивать двигательную активность, что ведет к детренированности, повышенной вероятности травматизации при физической нагрузке.
На фоне детренированности появляются мышечная слабость, повышенная утомляемость. Иногда после травм развиваются острые синовиты, типичными особенностями которых являются невоспалительный характер выпота и быстрое купирование симптоматики. Нередко беспокоят боли в спине. Интенсивность болевого синдрома может существенно варьироваться – от незначительных или умеренных болевых ощущений до упорных болей, существенно ограничивающих физические возможности пациента.
Поскольку СГС развивается на фоне врожденных нарушений синтеза белков, входящих в состав кожи, стенок сосудов, внутренних органов, у многих пациентов выявляются внесуставные проявления патологии. Типичными признаками являются чрезмерно растяжимая тонкая кожа, раннее развитие варикоза, появление синяков после незначительных травм. У некоторых больных обнаруживаются пролапс митрального клапана, опущение матки, почек, прямой кишки, грыжевые выпячивания различных локализаций.
Осложнения
У пациентов с СГС могут возникать повторные подвывихи суставов. Со временем боль начинает беспокоить даже при отсутствии значимых нагрузок, что связано с формированием менископатий, энтезопатий, бурситов, туннельных синдромов, других патологий. Отмечается повышенная вероятность раннего остеоартроза и остеохондроза. При манифестации вторичных поражений связочно-суставных структур жалобы видоизменяются, соответствуют клиническим проявлениям того или иного заболевания.
Наличие вегетативных нарушений, нередко выявляющихся при дисплазии соединительной ткани, утяжеляет течение болезни. На фоне постоянных артралгий, миалгий и дорсалгий, сочетающихся с обмороками, кардиалгиями, сердцебиением, ощущением нехватки воздуха, другими признаками вегетативной дисфункции, развиваются невротические расстройства.
Диагностика
Синдром гипермобильности суставов диагностируют врачи-ортопеды или ревматологи. Для выявления повышенной подвижности применяют так называемый счет Бейтона – совокупность признаков, которые обнаруживаются при проведении простых тестов, не требующих дополнительного оборудования:
- Переразгибание мизинца. Выпрямленный мизинец отклоняется к тыльной стороне кисти до образования угла 90 градусов.
- ПриведениеIпальца. Удерживая большой палец другой рукой, его можно подвести вплотную к предплечью.
- Переразгибание локтевого сустава. Предплечье отклоняется в сторону плеча до 10 и более градусов.
- Переразгибание коленного сустава. Угол между бедром и голенью составляет 10 или более градусов.
- Повышенная гибкость позвоночника. В положении стоя пациент может полностью положить ладони на пол, не сгибая колени.
За первые четыре положительных признака начисляется по 1 баллу с каждой стороны, за пятый – 1 балл. Наибольшее возможное количество баллов – 9. Используя результаты теста и данные анамнеза, выявляют СГС на основе определенных диагностических критериев:
1. Большие критерии:
- 4 и более балла по шкале Бейтона;
- жалобы на боли в 4 или более суставах на протяжении 3 или более месяцев.
2. Малые критерии:
- 1-3 балла по счету Бейтона для молодых пациентов и 0-2 балла для людей старше 50 лет;
- артралгии в 1-3 суставах или люмбалгии в течение 3 и более месяцев, спондилез, спондилолистез;
- наличие в анамнезе вывихов более чем в 1 суставе или повторных вывихов сустава;
- поражение околосуставных тканей: бурсит, тендинит, энтезопатия, эпикондилит и др.;
- высокий рост, худощавость, арахнодактилия, длинные ноги;
- изменения кожи: атрофии, чрезмерная растяжимость, стрии и т. д.;
- миопия либо нависающие веки;
- варикоз нижних конечностей, опущение внутренних органов, грыжи.
Для постановки диагноза необходимо 2 больших признака, 1 большой и 2 малых признака либо 4 малых признака. План обследования при подозрении на СГС определяется жалобами больных, может включать рентгенографию, КТ, МРТ различных сегментов, другие визуализационные методики. Для исключения ревматических болезней выполняют соответствующие лабораторные исследования.
Дифференциальная диагностика
Гипермобильность суставов, сочетающаяся с симптоматикой со стороны артикулярных, периартикулярных и мышечных тканей встречается при ряде других заболеваний, что обуславливает важность дифференциальной диагностики. В первую очередь СГС различают с синдромами Марфана и Элерса-Данлоса. В ряде случаев может потребоваться дифференцировка с синдромом Луиса-Дитца, несовершенным остеогенезом, иными патологиями.
Лечение синдрома гипермобильности суставов
Лечение СГС длительное, осуществляется в амбулаторных условиях. Центральное место в плане терапевтических мероприятий занимает коррекция образа жизни и режима физической активности. По показаниям немедикаментозные методы лечения дополняют лекарственной терапией.
Немедикаментозные методики
При применении физиологических способов нормализации состояния больных важны комплексный подход и раннее начало лечения. Особое значение немедикаментозные методы имеют в терапии пациентов младше 20 лет, у которых нередко удается существенно уменьшить проявления СГС в течение дальнейшей жизни. Рекомендуются:
- Оптимальный режим дня. Включает достаточное количество сна, чередование периодов работы и отдыха, регулярное полноценное питание. Важной частью режима является ежедневная умеренная физическая активность для профилактики детренированности.
- Коррекция двигательных стереотипов. Предусматривает отработку правильной техники движений. Осуществляется врачами ЛФК. Направлена на исключение нефизиологических нагрузок, предупреждение перегрузок, повторной травматизации.
- Кинезиотерапия. Разрабатываются комплексы статических и динамических упражнений для укрепления мышц в проблемных зонах. Комплексы ЛФК выполняются вначале со специалистом, затем – индивидуально в домашних условиях, по показаниям дополняются остеопатией, миофасциальным релизингом, механотерапией.
- Фиксация. Производится индивидуальный подбор ортезов. В ходе подбора учитываются уровень активности пациента, локализация болевого синдрома, выраженность изменений в различных анатомических зонах.
- Физиотерапия. Назначается в период обострений. Направлена на ликвидацию болевого синдрома, стимуляцию восстановления тканей. Применяются лекарственный электрофорез, лазеротерапия, магнитотерапия, другие методики.
Медикаментозные методы
В большинстве случаев для устранения болевого синдрома достаточно немедикаментозных мероприятий в сочетании с использованием местных средств (гелей, мазей) с отвлекающим и противовоспалительным эффектами. При недостаточной результативности показаны:
- Стимуляторы белкового обмена. В перечень рекомендованных средств входят кальцитонин, аскорбиновая кислота, никотиновая кислота, витамины группы В, минеральные комплексы, другие препараты, способствующие активизации образования коллагена, обеспечению баланса окислительно-восстановительных процессов.
- Корректоры обмена глюкозаминогликанов. Применяются медикаменты из группы хондропротекторов – глюкозамина сульфат, хондроитина сульфат и др.
- Стабилизаторы минерального обмена. Используются средства, содержащие различные формы витамина Д (эргокальциферол, альфакальцидол и аналоги), препараты кальция.
- Корректоры биоэнергетического состояния. Назначаются лекарства, в состав которых входят фосфолипиды и полиненасыщенные жирные кислоты (лецитин), рибоксин, мельдоний, незаменимые аминокислоты.
Медикаментозную терапию проводят несколько раз в год курсами продолжительностью около 2 месяцев с интервалом между курсами не менее 2-3 месяцев. Обычно курс включает по одному препарату из каждой группы. В начале следующего курса средства заменяют. Прием лекарств чередуют с физиотерапией. НПВС при синдроме гипермобильности суставов применять не рекомендуют из-за незначительной выраженности воспаления и возможного негативного влияния медикаментов на состояние соединительной ткани.
Прогноз
Прогноз благоприятный. При соблюдении рекомендованного режима активности, выработке правильных двигательных стереотипов большинство больных ведет обычный образ жизни, полностью сохраняет трудоспособность. В отдельных случаях возможно тяжелое течение с повторными эпизодами нетрудоспособности, необходимостью индивидуальной адаптации, иногда – вынужденной сменой профессии.
Профилактика
Первичная профилактика не разработана из-за врожденного характера патологии. Вторичные профилактические меры включают раннее выявление и регулярное наблюдение пациентов с гипермобильностью суставов, индивидуальный подбор физических нагрузок, профориентацию, разъяснение особенностей течения болезни для предупреждения невротических расстройств, создания настроя на необходимость коррекции в течение всей жизни.
1. Синдром гипермобильности суставов: клиническое значение, прогноз, взаимосвязь с риском возникновения остеоартроза/ Викторова И.А., Коншу Н.В., Румянцев А.В.// Архив внутренней медицины – 2015 - №2.
3. Гипермобильность суставов и гипермобильный синдром – клинические аспекты/ Шостак Н.А., Правдюк Н.Г., Тимофеев В.Т., Шеметов Д.А.// Неврология и ревматология – 2017 - №3.
4. Синдром гипермобильности суставов в ревматологии/ Сатыбалдыев А.М.// Современная ревматология – 2017 - №2.
Что такое синдром Марфана? Причины возникновения, диагностику и методы лечения разберем в статье доктора Боровиковой Ольги Игоревны, генетика со стажем в 7 лет.
Над статьей доктора Боровиковой Ольги Игоревны работали литературный редактор Маргарита Тихонова , научный редактор Сергей Федосов
Определение болезни. Причины заболевания
Синдром Марфана (Marfan; СМ) — генетически обусловленное заболевание, при котором происходит системное поражение соединительной ткани. [1]
Причины синдрома Марфана
Этиологией заболевания является мутация в гене FBN1 (фибриллина 1), расположенном в коротком плече пятнадцатой хромосомы в локусе 21.1. [2]
Тип наследования синдрома — аутосомно-доминантный. Для болезни характерна высокая пенетрантность (частота появления гена) и различная экспрессивность. [5]
Соотношение представителей мужского пола и женского одинаковое.
При обнаружении схожих симптомов проконсультируйтесь у врача. Не занимайтесь самолечением - это опасно для вашего здоровья!
Симптомы синдрома Марфана
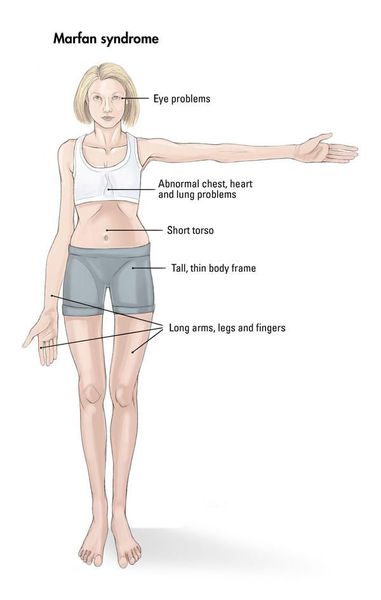
Наблюдается постоянно прогрессирующее развитие заболевания. У новорожденных детей выявляются удлинённые тонкие пальцы на верхних и нижних конечностях и удлинённые тонкие конечности (долихостеномелия). [1] У таких пациентов, помимо долихостеномелии, отмечается:
- повышенное физическое развитие;
- недостаток веса;
- удлинённый череп;
- вытянутое лицо;
- арахнодактилия (аномально удлинённые узкие пальцы);
- слабость и недоразвитие мышечной системы и жировой клетчатки;
- неловкие движения. [3]
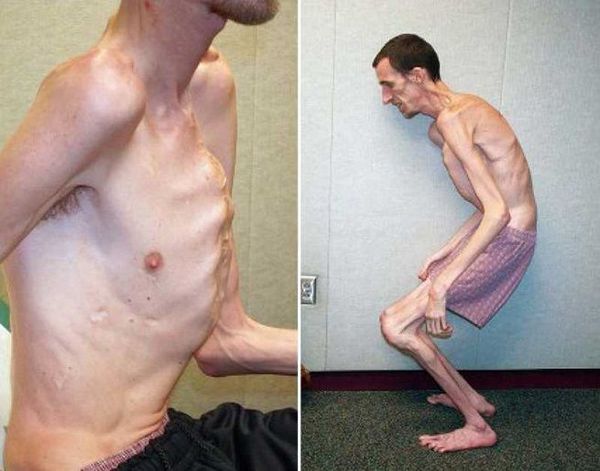
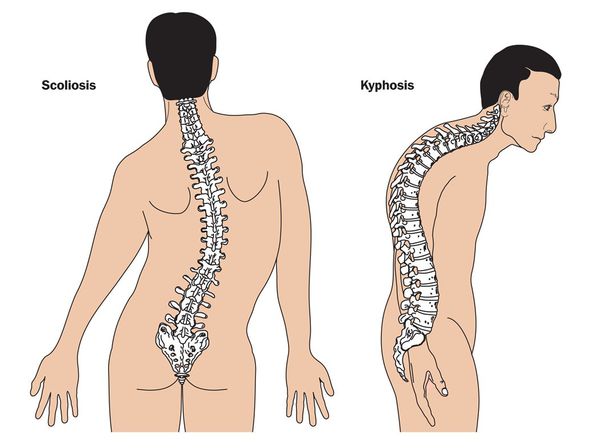
Кожа имеет повышенную растяжимость, разболтанные суставы. У большинства больных наблюдается высокое аркообразное нёбо, изменения формы грудной клетки (воронкообразная, килевидная) и искривления позвоночника (сколиоз в 60%, кифоз (изгиб позвоночника с образованием горба), ювенильный остеохондроз), уплощение свода стопы, аускультативные признаки порока сердца (шумы). [4] Длина третьего пальца руки — 10 см и больше (скрининговый тест у детей 7-18 лет): возрастает соотношение размаха верхних конечностей к длине тела.
Офтальмологические симптомы (близорукость, подвывих хрусталика в 75% случаев, его округлость или гипоплазия, отслойка сетчатки) и астенические признаки (усталость, вялость) обращают на себя внимание со второго года жизни, изменения формы грудной клетки появляются в возрасте старше четырёх лет, патология сердца и сосудов выявляется в дошкольном возрасте. [1]
Почти у всех больных выявляются пороки сердца и аорты. Часты бедренные и паховые грыжи, поражение клапанов в венах, их варикозное расширение, геморрагический синдром, рецидивирующие вывихи, поражение лёгочной системы (самопроизвольный пневмоторакс, эмфизематозное расширение лёгких), опущение почек. [2]
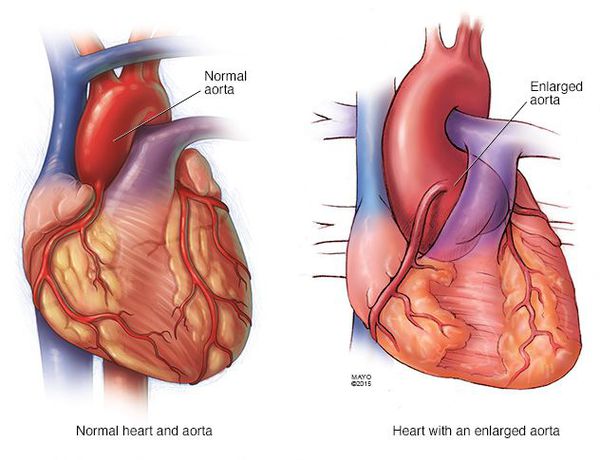
По данным многих исследований, абсолютное большинство больных с синдромом Марфана отмечают ухудшение эмоционального фона, утрату чувства радости и увлечённости профессиональной деятельностью, частую смену настроения, повышенную возбудимость, чувство тревоги. Результатом этого является снижение социальной активности, ухудшение качества жизни и значительное уменьшение социальной адаптации. [3]
У таких пациентов часто наблюдается трахеобронхиальная дискинезия (нарушение дыхательной системы) за счёт слабости соединительнотканного каркаса бронхов. Это проявляется рецидивирующими воспалительными заболеваниями бронхолегочной системы, обструктивными нарушениями, бронхиальной астмой, эмфиземой лёгких (повышенное содержание воздуха в лёгочной ткани). [4] Встречаются осложнения, которые проявляются скоплением воздуха в грудной клетке, сопровождающиеся сдавлением лёгких и средостения (срединной области грудной клетки), подкожной эмфиземой. Наблюдается неадекватный ответ на бронхолитики. Обструктивные явления (непроходимость) затрагивают преимущественно верхние отделы респираторного тракта. [3]
Описаны характерные изменения на электрокардиограмме, включающие синдром раннего возбуждения желудочков, преждевременные желудочковые комплексы, нестабильность конечной части желудочкового комплекса в задненижних отведениях. [3]
Патология ритма чаще всего проявляются блокадой правой ножки пучка Гиса или смешанной экстрасистолией. [6]
У больных синдромом Марфана с патологией ритма сердечной деятельности и проводимости синдром вегетативной дисфункции чаще протекает по ваготоническому типу, в виде пресинкопальных, обморочных и астеновегетативных состояний, болезненных ощущений в области сердца, цефалгии напряжения (головной боли) и зачастую сочетается с психопатологическими расстройствами. [4]
Органы пищеварения также задействованы в патологическом процессе, что проявляется дискинезией (нарушением моторики) билиарного тракта со снижением моторики гладкомышечной мускулатуры, недостаточностью кардии, грыжевыми выпячиваниями пищеводного отверстия диафрагмы, аномалиями желчевыводящих протоков, долихосигмой (увеличением сигмовидной кишки), хроническим гастродуоденитом (воспалением слизистой желудка и двенадцатиперстной кишки), дисбиозом (нарушением нормальной микрофлоры) кишечника, изменениями поджелудочной железы. [3]
У пациентов с синдромом Марфана чаще, чем у здоровых людей, встречаются приобретённые аномалии почек: повышенная подвижность почек, нефроптоз (опущение почки), пиелоэктазии (аномальное расширение лоханок), повышена частота удвоения почек.
Патогенез синдрома Марфана
Более половины веса человека представлено соединительной тканью, из неё состоит наша главная опора — скелет, внешние покровы — кожа. Сосуды, кровь и лимфа тоже состоят из соединительной ткани.
К клеткам соединительной ткани относятся фибробласты и их разновидности (остеобласты, хондроциты, одонтобласты, кератобласты), макрофаги (гистиоциты) и тучные клетки (лаброциты). [7]
Мезенхима — проводник конституциональных, генетических и эпигенетических составляющих жизни человека. Патология соединительной ткани детерминирует определенное патологическое действие на весь организм в целом, на его физиологию и его конституциональные особенности. [3]
При болезни Марфана происходит замена нуклеотидов в гене, содержащем информацию о структуре пептида фибриллина-1. Этот белок относится к гликопротеидам, принимает участие в микрофибриллярном комплексе, он обеспечивает основу эластических фибрилл соединительной ткани.
Межклеточный матрикс позволяет соединительной ткани поддерживать постоянную структуру, в нём находится огромное количество факторов роста, которые обеспечивают постоянное обновление клеток.
В крупных сосудах, связочном аппарате содержится большое количество эластиновых фибрилл, поражение которых и даёт основные клинические проявления синдрома Марфана.
При синдроме Марфана значительно поражается трансформирующий фактор роста бета (TGF-β), нарушается связывание его неактивной формы, что приводит к повышению биоактивности данного фактора, с чем связано появление многих проявлений болезни. [4]
Патология фибриллина приводит к патологии формирования волокон, что вызывает утерю прочности и эластичности кожи и других соединительнотканных структур.
Изменение структуры коллагеновых волокон приводит к нарушению первичного звена гемостаза у пациентов с синдромом Марфана. [6]
Имеются данные о дефектах мембранных и цитоплазматических механизмов проведения сигнала непосредственно в самом тромбоците, приводящих к нарушениям агрегации (объединения). Показано наличие самостоятельного мембранного дефекта тромбоцитов, протекающего с нарушением реакций высвобождения и транспорта внутриклеточного кальция. [6]
Эластические фибриллы имеют вполне определенные механизмы участия в системе гемостаза. В сосудах с низкой скоростью сдвига происходит адгезия («прилипание») тромбоцитов к эластину через фибронектин. [7] Регистрируется снижение его уровня в крови у людей с синдромом Марфана. Фибронектин, в свою очередь, образуется в клетках эндотелия и участвует в последующих репаративных процессах, создавая основу для производства других компонентов соединительной ткани — фибробластов. [4] Таким образом, совершенно неоспоримо участие сосудистой стенки в реакциях свертываемости крови, и неизбежен вывод о возможных патологиях протекания нормальных гемостатических процессов при изменении состояния её структурных компонентов и процессов сосудистой регуляции.
Отмечена роль гормонального дисбаланса в развитии и усугублении дефектов соединительнотканных структур. [3]
Тромботические проявления детерминированы нарушением реологии (вязкости) крови в патологически извитых сосудах брахиоцефальной зоны. [3]
Поражение желудочно-кишечного тракта детерминировано тем, что эта система богата коллагеном. Наблюдаются дискинезия билиарного тракта по гипомоторному типу, грыжи пищеводного отверстия диафрагмы, аномалии желчных путей, долихосигма, хронический гастродуоденит со стёртой клинической картиной, склонностью к торпидному течению. [3]
Классификация и стадии развития синдрома Марфана
Код синдрома Марфана по Международной классификации болезней (МКБ-10): Q87.4.
- стёртая (поражено не более двух систем, изменения выражены незначительно);
- выраженная (незначительные изменения в трёх системах либо значительное поражение одной и более систем).
Выделяют различные типы по степени тяжести:
Частота тяжёлых форм — 1 к 25000-50000 (при общей частоте диагностированных случаев 1 к 10000-15000).
По характеру течения:
- прогрессирующая форма;
- стабильная форма.
Чаще всего первые признаки синдрома Марфана проявляются еще в детском периоде, с возрастом происходит прогрессирование симптомов, усиление клинических проявлений.
Осложнения синдрома Марфана
К самым частым осложнениям синдрома Марфана относятся:
- Снижение зрения, вплоть до слепоты, обусловленное слабостью цинновой связки (ресничного пояска) и подвывихом, вывихом хрусталика. [7]
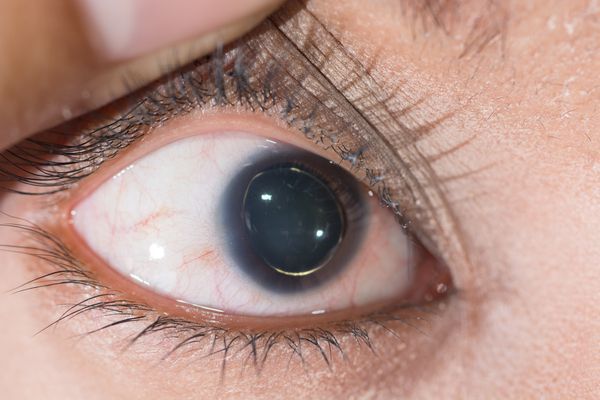
- Сердечная недостаточность по застойному типу, обусловленная нарушением сократимости сердечной мышцы, недостаточностью митрального клапана. [6]
- Разрывы крупных сосудов, связанные с дилатацией (расширением), истончением стенки сосудов. Чаще всего происходит поражение аорты (в основном из-за изменения гемодинамики при беременности). [7]
- Расслаивающая аневризма аорты, приводящая к смерти больных.
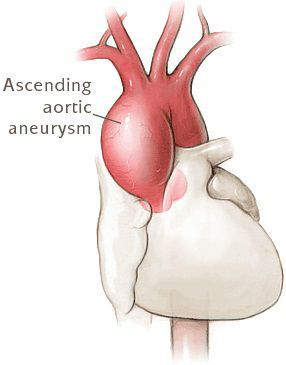
Диагностика синдрома Марфана
Диагностика Синдрома Марфана основывается на клинических данных, выявлении изменений в гене FBN1. [5]
Часто при сборе генеалогического анамнеза выявляются родственные случаи со скрытым течением заболевания. [1]
Способы обнаружения арахнодактилии: [3]
- Симптом Steinberg (признак первого пальца). Первый палец виден из-под hypothenar при напряжённом кулаке.
- Симптом Walker-Murdoch (признак запястья). При обхватывании кистью в области лучезапястного сочленения контралатеральной верхней конечности первый палец заходит за пятый.
- Определение пястного индекса. Определяется при помощи рентгенографии. Средняя длина пясти, делённая на усреднённую ширину отрезка от второй до четвертой пястной кости. При нормальном соотношении этот показатель соответствует 5,4-7,9, в то время, как при синдроме Марфана — больше 8,4.
В 2010 году группа специалистов систематизировала международные Гентские критерии для верификации синдрома Марфана. Верификация зависит от данных генеалогического анамнеза. [3]
При отсутствии генеалогического анамнеза:
- увеличение диаметра аорты >, = 2 ϭ + эктопия хрусталика = СМ;
- увеличение диаметра аорты >, = 2 ϭ + выявленные изменения в гене FBN1 = CM;
- увеличение диаметра аорты >, = 2 ϭ + >, = 7 системных признаков = СМ;
- эктопия хрусталика + наличие изменений в гене FBN1 + дилатация аорты = СМ;
При наличии генеалогического анамнеза:
- Эктопия хрусталика + случай СМ в семье = СМ;
- >, = 7 системных проявлений + случай СМ в семье = СМ;
- увеличение диаметра аорты >, = 2 ϭ + случай СМ в семье = СМ.
В пятнадцати процентах появление ребёнка с синдромом Марфана спорадическое (случайное), у родителей могут быть слабые проявления. У родственников пациентов встречаются заболевания желудочно-кишечного тракта, поражения позвоночника, заболевания глаз. [3]
При малейшем подозрении на синдром Марфана необходима консультация офтальмолога. В анализе мочи таких пациентов отмечается повышение уровня оксипролина, гликозаминогликанов, но эти показатели низкоспецифичны, могут быть при различных дисплазиях соединительной ткани. Выделение оксипролина является показателем тяжести заболевания. Наблюдается нарушение свертываемости крови на тромбоцитарном уровне. [3]
Оценка системных признаков вовлечённости соединительной ткани
Синдром Элерса-Данлоса (Ehlers-Danlos) - синонимы, авторы, клиника
Синонимы синдрома Элерса-Данлоса. S. Danlos. S. Meekrin—Ehlers—Danlos. Эластическая фибродисплазия. Генерализованная эластическая фибродисплазия. Мезодермальная врожденная дистрофия. Врожденная множественная слабость суставов. Cutis hyperelastika dermatorrhexis. Эластическая кожа. Гиперэластическая кожа. «Каучуковый человек». Мезенхимоз (Leger).
Определение синдрома Элерса-Данлоса. Врожденная мезенхимальная дисплазия с клиническими проявлениями поражения соединительной ткани кожи, мышц и связочного аппарата суставов.
Симптоматология синдрома Элерса-Данлоса:
1. Гиперэластичность и повышенная растяжимость кожи с развитием атрофических рубцов и очагов гиперпигментации.
2. Повышенная ломкость кровеносных сосудов кожи; рецидивирующие гематомы, кровотечения.
3. Недостаточное развитие подкожножировой ткани.
4. Патологическая слабость и чрезмерная растяжимость капсульного и соединительнотканного аппарата суставов. Гиперрастяжимость суставов, подвывихи, вывихи с последующим развитием вторичных деформаций.
5. Гипотония скелетной мускулатуры.
6. Иногда неврологические расстройства.
7. Нередко наблюдают другие дегенеративные стигматы и врожденные пороки развития (выступающий лобный бугор, гипертелоризм, сколиоз, вывих тазобедренного сустава, синдактилия, бронхоэктазии, снижение интеллекта, хондродистрофия — S. Parrot и др.).
8. Чаще болеют мужчины (11:5).
Этиология и патогенез синдрома Элерса-Данлоса. Наследственное страдание, по-видимому, с мономерной доминантной передачей. Встречаются, однако, формы, при которых не удается установить роли наследственных факторов (новые мутации?). Патогенетически речь идет о системной мезодермальной дисплазии с порочным развитием коллагеновых волокон и, возможно, с нарушением обмена мукополисахаридов.
Дифференциальный диагноз синдрома Элерса-Данлоса. Chalacoderma.
Синдром Элерса-Данло.
У ребенка наблюдается ряд классических признаков заболевания, к которым относятся (а) повышенная склонность к кровоподтекам,
(б) гиперэластичность кожи и (в) гиперрастяжимость суставов.
У отца ребенка (г) множественные обширные атрофические лиловые рубцы на коленях накопились за многие годы в результате травм.
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021

Синдром Элерса-Данлоса – это наследственное заболевание, связанное с патологией соединительной ткани. При этой патологии наблюдается повышенная растяжимость кожи. Такое явление специалисты связывают с нарушением структуры коллагена. В основе нарушений синтеза этого вещества, составляющего одну из наибольших частей соединительной ткани организма человека, лежат мутации в генах, кодирующих этот процесс. Заболевание наследуется по аутосомно-доминантному или аутосомно-рецессивному принципу. В первом случае синдром Элерса-Данлоса скорее всего напрямую передастся от родителя ребенку. В втором – такая вероятность в разы меньше.
Синдром Элерса-Данлоса – Виды
Выделяют несколько типов заболевания:
- Классический – характеризуется разболтанностью суставов, вялостью кожных покровов. Нередко у пациентов еще в детском возрасте формируется сколиоз. Суставы нестабильны и легко деформируются. Заболевание передается по наследству, тип передачи патологии аутосомно-доминантный;
- Гиперподвижность – наиболее характерный симптом – гипермобильность суставов. Тип наследования аутосомно-доминантный;
- Сосудистый – среди признаков заболевания на первый план выходит хрупкость сосудов, проявляющаяся спонтанными разрывами артерий. Это приводит к частым внутренним кровотечениям, которые могут составлять угрозу для жизни больного; – для этой формы заболевания наиболее характерно появление такой деформации позвоночника как сколиоз. Также нередко отмечаются изменения в глазном яблоке;
- Артроклазия – у больных с такой формой заболевания наблюдается маленький рост и повышенная мобильность суставов ;
- Дерматоспраксис – характеризуется своеобразным внешним видом кожи, которая становится вялой, обвисает.
Синдром Элерса-Данлоса – Симптомы
Для синдрома Элерса-Данлоса характерны такие клинические признаки:
- Большая подвижность в суставах, что обуславливает возможность выгибать конечности неестественным образом. Так, мизинец у таких больных может сгибаться более чем на 90 0 , а большой палец легко приводится к предплечью. Локтевой сустав способен переразгибаться на 10 и более градусов. Нередко первые признаки такого синдрома обнаруживаются у детей в школьном возрасте. На занятиях по физкультуре дети с синдромом Элерса-Данлоса проявляют необычную гибкость, свободно дотягиваясь ладонями до пола в наклоне вперед. Большой процент пациентов с таким диагнозом обнаруживается среди гимнасток и некоторых других спортсменов. К сожалению, нередко при таких занятиях могут возникать вывихи, растяжения связок. У таких детей часто обнаруживается плоскостопие;
- Из-за общей патологии соединительной ткани определенные нарушения наблюдаются и в офтальмологической сфере. Из-за растяжения апоневроза мышцы, поднимающей верхнее веко, у таких пациентов может наблюдаться птоз (опущение верхнего века). Нередко наблюдается отслойка сетчатки;
- Кожа у таких больных легко повреждается, она очень гладкая и эластичная. Кожные сосуды легко повреждаются и кровоточат. При малейшем воздействии могут появляться синяки и кровоподтеки. При заживании даже небольшие ранки оставляют рубцы. Нередко наблюдаются стрии или растяжки из-за натяжения кожи в области поясницы. В послеоперационном периоде у таких больных нередко развивается такое осложнение как расхождение швов, что связано с плохим заживанием ран и разрезов кожи.
- У больных с таким синдромом чаще, чем у других людей, наблюдаются вывихи, растяжения связок;
- У больных наблюдается деформация грудной клетки различного характера. Может формироваться сколиоз, лордоз, кифоз, уплощение позвоночника, а также вдавление грудины;
- У пациентов с синдромом Элерса-Данлоса часто наблюдаются грыжи различной локализации. Это связано с ослаблением передней стенки живота из-за патологии коллагенообразования;
- Среди стоматологической патологии можно назвать развитие парадонтоза, множественного кариеса;
- На нижних конечностях нередко формируется варикозное расширение вен, ведь соединительная ткань участвует и в поддержании тонуса венозной стенки;
- Недостаточная прочность сосудистой стенки приводит также к формированию аневризм. Особенно опасно, если они локализуются в сосудах головного мозга, что может привести к внутричерепному кровоизлиянию;
- Из-за слабости связочного аппарата может происходит опущение внутренних органов (матки, желудка);
- Изменения могут затрагивать и сердце, приводя к пролапсу митрального клапана.
Синдром Элерса-Данлоса – Диагностика в Израиле
Диагностика синдрома Элерса-Данлоса основывается на обследовании больного. При этом изучается состояние его опорно-двигательного аппарата, оценивается подвижность суставов, состояние кожи. Кроме этого, полезным будет проведение следующих исследований:
- Биопсия кожи – после забора образцов кожного покрова они доставляются в лабораторию, где изучается микроскопический состав кожи;
- Ультразвуковое исследование внутренних органов – необходимо для выявлении патологии, связанной с недостаточность связочного аппарата;
- Консультация офтальмолога – при проведении биомикроскопии, фундус-графии, компьютерной томографии глазного яблока можно выявить патологию и предупредить дальнейшее ее прогрессирование.
Синдром Элерса-Данлоса – Лечение в Израиле
Этиологического лечения заболевания на сегодняшний день не существует. Лечение, как правило, симптоматическое. Израильские хирурги успешно проводят операции, которые помогают укрепить суставы у пациентов с синдромом Элерса-Данлоса. Стабилизация сустава достигается при помощи имплантации синтетических или аутологичных материалов таким образом, чтобы предотвратить избыточное переразгибание конечности. Операции, проводимые на внутренних органах, помогают бороться с их опущением. Сосудистые хирурги успешно проводят эндоскопические операции, направленные на выключение аневризм из кровотока или укрепление их стенки, что предотвращает их разрыв.
Израильская медицина имеет прекрасные возможности в лечении проявлений синдрома Элисона-Данлоса!
Внимание! Все поля формы обязательны. Иначе мы не получим Вашу информацию. Альтернативно пользуйтесь [email protected]
Лечение в клиниках “АССУТА” и “ХАДАССА”
Вас интересует лечение в Израиле?
Крупнейшие профессиональные больницы Израиля – «Ассута» в Тель-Авиве и «Хадасса» в Иерусалиме предлагают реальную возможность получить качественное и специально для вас подобранное лечение у замечательных специалистов по адекватным ценам.
Мы помогаем найти решение ваших проблем со здоровьем, а также предоставляем полную информацию о лучших израильских врачах.
Читайте также: