
Что такое лимфопролиферативное заболевание кожи
Обновлено: 19.04.2024
Информацию из данного раздела нельзя использовать для самодиагностики и самолечения. В случае боли или иного обострения заболевания диагностические исследования должен назначать только лечащий врач. Для постановки диагноза и правильного назначения лечения следует обращаться к Вашему лечащему врачу.
Лимфомы: причины появления, симптомы, диагностика и способы лечения.
Определение
Лимфомы представляют собой группу злокачественных опухолевых заболеваний лимфатической ткани, которая в нормальных условиях отвечает за иммунитет. Одним из первых клинических симптомов лимфом является увеличение лимфатических узлов разных локализаций.
Лимфомы, как и другие злокачественные опухоли, обладают склонностью к метастазированию, то есть миграции опухолевых клеток через кровь и лимфу, а также контактным путем в интактные (здоровые) ткани с последующим размножением в них.
Практически все виды лимфогранулематоза, или лимфомы Ходжкина, и около 65% неходжскинских лимфом проявляются безболезненным увеличением лимфатических узлов - 2 см и более. Однако диагноз того или иного вида лимфомы устанавливают исключительно на основании морфологического исследования биопсийного материала, взятого из опухоли.
Заболеваемость лимфомой Ходжкина в России составляет около 2 случаев на 100 тыс. населения в год, а неходжкинскими лимфомами – 5–7 случаев на 100 тыс.
Причины появления лимфом
Существует несколько возможных причин развития лимфом. Важно понимать, что в основе развития любого злокачественного заболевания, в том числе и лимфом, лежит нарушение функций клеток.
Способность к неконтролируемому росту опухоли возникает при нарушении естественного процесса деления клетки: в норме клетка делится ограниченное количество раз, созревает, выполняет определенную функцию, после чего заканчивает свою жизнь.
Для злокачественных клеток характерно нарушение процессов созревания и дифференцировки (процесса, в ходе которого клетка приобретает специфические особенности и способность выполнять определенные функции). У клеток появляются новые функции – они начинают продуцировать белки и токсины, возникает способность «ускользать от иммунного надзора», в результате чего меняется процесс естественной гибели клетки (апоптоза).
Некоторые вирусы могут напрямую влиять на ДНК лимфоцитов и способствовать их превращению в злокачественные клетки.
Лимфоциты делятся на два подвида: Т- и В-клетки (Т- и В-лимфоциты). Заражение Т-клеточным лимфотропным вирусом человека (HTLV-1) повышает риск возникновения у пациента определенных типов Т-клеточной лимфомы. Вирус передается половым путем и через кровь, а также от матери к ребенку через грудное молоко.
Вирусы, ослабляющие иммунологический надзор, косвенно являются причиной развития некоторых типов лимфом.
Некоторые инфекции могут стать причиной развития лимфомы из-за постоянной активации иммунной системы. При хронических инфекциях вырабатывается огромное количество лимфоцитов, что повышает вероятность возникновения мутаций во время деления клеток и ведет к риску формирования лимфомы. К подобным инфекциям относят Helicobacter pylori (часто встречается при хроническом гастрите и язвенной болезни желудка и двенадцатиперстной кишки), Chlamydophila psittaci (риск развития лимфомы в тканях глаза), вирусы гепатита В и С при длительном инфицировании.
К неинфекционным факторам риска развития лимфом относят:
- Воздействие химических веществ. Многочисленные исследования доказали, что влияние гербицидов, инсектицидов, бензола связано с повышенным риском развития лимфом.
- Наличие аутоиммунных заболеваний (ревматоидного артрита, системной красной волчанки и т.д.) является значимым фактором риска развития лимфом.
- Радиационное облучение оказывает большое влияние на скорость возникновения мутаций клеток и их озлокачествление.
- Генетические факторы. Наличие близкого родственника, страдающего лимфомой, значительно увеличивает риск.
- Врожденная недостаточность иммунитета при некоторых заболеваниях.
- Возраст. Некоторые типы лимфом развиваются у детей и подростков, а другие, напротив, у лиц старше 60 лет.
- Грудные импланты в редких случаях способствуют развитию одного из типов крупноклеточной лимфомы молочных желез.
Для лимфомы Ходжкина характерно наличие специфических клеток Рид–Березовского–Штернберга в пораженных лимфатических узлах, селезенке, печени или костном мозге.
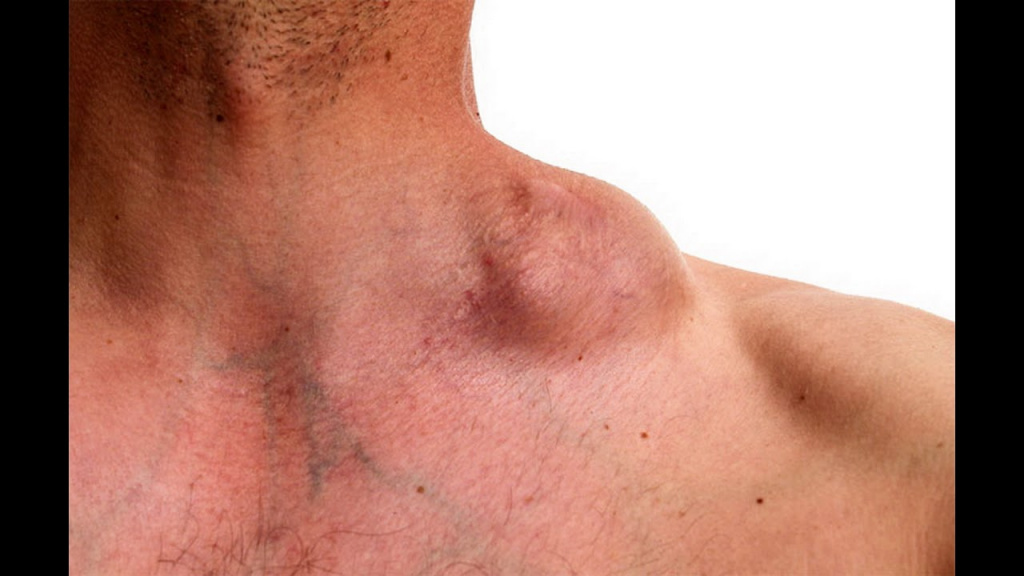
Увеличенные шейные лимфоузлы у пациента с лимфомой Ходжкина
К группе неходжкинских лимфом относятся:
- фолликулярная лимфома;
- лимфома маргинальной зоны;
- диффузная крупноклеточная В-клеточная лимфома;
- лимфома из клеток мантии;
- лимфома Беркитта;
- первичная лимфома центральной нервной системы;
- нодальные Т-клеточные лимфомы;
- первичные кожные лимфомы;
- неходжкинские лимфомы у больных, инфицированных ВИЧ;
- неходжкинские лимфомы у больных, инфицированных вирусами гепатита В и С.
Самый ранний симптом – увеличение лимфатических узлов на фоне полного благополучия. Увеличенные лимфоузлы безболезненны.
Различные клинические проявления зависят от локализации опухоли: если происходит рост опухоли в лимфатических узлах грудной клетки, возможно сдавливание дыхательных путей и появление охриплости голоса, нарушение глотания, затруднение дыхания, навязчивый кашель.
При расположении лимфомы в средостении появляется синдром сдавления верхней полой вены: возникают головные боли, отек и расширение вен лица и шеи.
В некоторых случаях присутствуют только общие симптомы: потеря аппетита, снижение массы тела, повышение температуры тела, ночная потливость, слабость, иногда – кожный зуд и боль в увеличенных лимфоузлах.
На фоне снижения иммунитета могут присоединяться инфекционные заболевания - часто активируется вирус ветряной оспы, проявляющийся опоясывающим герпесом.
Описаны случаи поражения органов брюшной полости (проявляется увеличением объема живота за счет роста опухоли и скопления жидкости, появлением болей, желтухи, тошноты и рвоты) и костного мозга (боли в костях, спине, частые переломы, бледность, повышенная кровоточивость).
Диагностика лимфом
Диагноз того или иного вида лимфомы устанавливают исключительно на основании морфологического исследования биопсийного материала (кусочка или целого лимфоузла, полученного в ходе небольшой операции), взятого из опухоли (гистологические, цитогенетические и молекулярно-генетические методы исследования). Дополнительные исследования врач назначит для диагностики стадии заболевания и его осложнений. Список исследований зависит от предположительной локализации опухоли и может быть изменен.
-
Клинический анализ крови.
Синонимы: Общий анализ крови, ОАК. Full blood count, FBC, Complete blood count (CBC) with differential white blood cell count (CBC with diff), Hemogram. Краткое описание исследования Клинический анализ крови: общий анализ, лейкоформула, СОЭ См. также: Общий анализ – см. тест № 5, Лейкоцит.
ОПРЕДЕЛЕНИЕ
Первичные лимфомы кожи (ЛК) представляют собой гетерогенную группу заболеваний, включающую кожные Т- и В-клеточные лимфомы, которые возникают в коже без обнаружения внекожных мест локализации на момент постановки диагноза. Первичные ЛК занимают второе по частоте встречаемости место среди экстранодальных неходжкинских лимфом (после лимфом желудочно-кишечного тракта) и представляют собой отдельный клинический и гистопатологический подтип экстранодальных лимфом, часто отличаясь от соответствующих им нодальных гистологических аналогов не только по характеру течения и прогнозу, но и по наличию специфических хромосомных транслокаций и экспрессии различных онкогенов.
СИНДРОМ СЕЗАРИ
Шифр по Международной классификации болезней МКБ-10
С84.1
Синдром Сезари (СС) представляет собой Т-клеточную лимфому кожи, характеризующуюся эритродермией, генерализованной лимфаденопатией и наличием циркулирующих злокачественных Т-лимфоцитов (клеток Сезари).

Автоматизация клиники: быстро и недорого!
- Подключено 300 клиник из 4 стран
- 800 RUB / 4500 KZT / 27 BYN - 1 рабочее место в месяц

Автоматизация клиники: быстро и недорого!
- Подключено 300 клиник из 4 стран
- 1 место - 800 RUB / 4500 KZT / 27 BYN в месяц
Мне интересно! Свяжитесь со мной
Классификация
В 2005 году Всемирная организация здравоохранения совместно с Европейской Организацией по изучению и лечению рака (European Organization for Research and Treatment of Cancer – EORTC) создали ВОЗ-EORTC классификацию ЛК, которая содержит наиболее полную клиническую, гистологическую, иммунофенотипическую, молекулярно-биологическую и генетическую информацию, касающуюся первичных ЛК и нескольких лимфопролиферативных заболеваний, часто начинающихся с поражения кожи.
ВОЗ-EORTC-классификация кожных лимфом
В-клеточные лимфомы кожи
Гемодермии из клеток-предшественников
Эпидемиология
СС составляет менее чем 5% от всех первичных кожных лимфом. Болеют преимущественно люди пожилого возраста с преобладанием пациентов мужского пола, средний возраст начала заболевания составляет 60-65 лет.
Клиническая картина
Cимптомы, течение
СС начинается с развития эритродермии, которая сопровождается сильным зудом и шелушением. Впоследствии присоединяется ладонно-подошвенный гиперкератоз, алопеция и ониходистрофия. Увеличение периферических лимфатических узлов может происходить за счет дерматопатической лимфаденопатии или вовлечения их в опухолевый процесс. При лабораторных исследованиях обнаруживается поражение периферической крови.
Диагностика
Международным обществом по лимфомам кожи (ISCL) и Европейской организацией по изучению и лечению рака (EORTC) выработаны следующие критерии диагностики СС:
- отсутствие предшествующего ГМ;
- генерализованная эритродермия (диффузная эритема, покрывающая не менее 80% поверхности тела с/без шелушения);
- наличие в крови доминантного клона Т-лимфоцитов (определяется при помощи ПЦР или Southern blot);
- наличие одного или более следующих признаков:
1) абсолютное количество клеток Сезари в крови ≥ 1000 клеток/мм 3 ;
2) повышено содержание CD3+ или CD4+ клеток периферической крови с коэффициентом отношения CD4/CD8 ≥ 10 (определяется при помощи проточной цитометрии);
3) повышено содержание CD4+ клеток периферической крови с аберрантным иммунофенотипом, включающем отсутствие экспрессии CD7 (≥ 40% CD4+СD7- клеток) или CD26 (≥ 30% CD4+CD26- клеток).
Гистологическое, иммуногистохимическое и молекулярно-биологическое (определение реарранжировки гена Т-клеточного рецептора методом ПЦР) исследования кожи и лимфатических узлов (в случае их увеличения ≥ 1,5 см) являются дополнительными методами диагностики в неясных диагностических ситуациях у пациентов с хронической эритродермией неясной этиологии.
Стадирование и план обследования
Стадирование СС проводится согласно пересмотренной TNM классификации, предложенной Международным обществом по лимфомам кожи и Европейской организацией по изучению и лечению рака (ISCL-EORTC staging system) (см. раздел «Грибовидный микоз»). Так как пациенты с СС характеризуются эритродермией (Т4) и В2–вовлечением крови, они расцениваются как имеющие IVA или IVB стадию заболевания.
Для пациентов с СС рекомендован следующий план обследования:
- при наличии узлов – определение их общего количества, размеров наибольшего узла и вовлеченных областей кожи;
- идентификация пальпируемых лимфатических узлов и органомегалии при физикальном осмотре;
- определение абсолютного количества клеток Сезари в крови, проточная цитометрия (включая CD4+СD7- и CD4+CD26-), определение реарранжировки гена ТКР в крови;
- клинический и биохимический анализ крови (ЛДГ, мочевая кислота (маркеры агрессивного течения заболевания), печеночные ферменты);
- определение маркеров вируса гепатита В (HbsAg, anti-HbcAg, anti-HbsAg), гепатита С, ВИЧ и HTLV-1;
- УЗИ периферических лимфатических узлов;
- компьютерная томография органов грудной клетки, брюшной полости и малого таза;
- биопсия кожи (гистологическое исследование, иммуногистохимическое исследование, включающее следующие маркеры: CD2, CD3, CD4, CD5, CD7, CD8, CD20, CD30, определение реарранжировки гена ТКР);
- биопсия лимфатических узлов > 1,5 см в диаметре и/или с плотной, неравномерной консистенцией (гистологическое исследование, проточная цитометрия, определение реарранжировки гена ТКР);
*выполняется врачами-специалистами с хирургической специализацией. При выявлении специфического поражения лимфатических узлов пациент передается для дальнейшего ведения и лечения гематоонкологам.
- трепанобиопсия костного мозга.
*выполняется врачами-специалистами, владеющими методикой забора трепанобиоптата костного мозга (хирургами или гематоонкологами). При выявлении специфического поражения костного мозга пациент передается для дальнейшего ведения и лечения гематоонкологам.
Дополнительно может использоваться магнитно-резонансная и позитронно-эмиссионная томография.
Дифференциальный диагноз
СС необходимо дифференцировать от других видов эритродермических кожных Т-клеточных лимфом и эритродермий другой этиологии:
1) Эритродермическая форма ГМ (Э-ГМ): эритродермическая кожная Т-клеточная лимфома, развившаяся на фоне течения ГМ с отсутствием вовлечения крови. При развитии поражения крови и наличии выше перечисленных диагностических критериев для СС такие случаи рекомендовано обозначать как «СС с предшествующим ГМ» или «вторичный СС».
2) Эритродермическая кожная Т-клеточная лимфома, другая: случаи, которые не удовлетворяют диагностическим критериям СС и Э-ГМ.
3) Доброкачественные воспалительные дерматозы, характеризующиеся эритродермией и повышением количества клеток Сезари в периферической крови (например, актинический ретикулоид или синдром лекарственно-индуцированной псевдолимфомы). Если абсолютное количество клеток Сезари в крови ≥ 1000 клеток/мм 3 или коэффициент отношения CD4/CD8 ≥ 10, такие случаи рекомендовано обозначать как «псевдо-СС».
Для дифференциальной диагностики различных видов эритродермических состояний, перечисленных выше, рекомендуется следующий алгоритм.

Алгоритм дифференциальной диагностики эритродермической кожной Т-клеточной лимфомы, синдрома Сезари и доброкачественных эритродермий
ТКР – Т-клеточный рецептор
Э-КТКЛ – эритродермическая кожная Т-клеточная лимфома
*Анормальный фенотип: повышенная популяция CD4+ клеток в периферической крови; аберрантный фенотип: отсутствие экспрессии CD7 (≥ 40% CD4+СD7- клеток) или CD26 (≥ 30% CD4+CD26- клеток)
Лечение
Цели лечения: достижение полной (частичной) ремиссии с последующим контролем течения заболевания.
Общие замечания по терапии
Различными исследованиями было показано, что факторами, влияющими на прогноз заболевания, являются:
- возраст пациента;
- повышение уровня ЛДГ в крови;
- поражение лимфатических узлов;
- степень тяжести вовлечения крови.
Выбор вида терапии при СС должен базироваться на определении степени тяжести заболевания (учитываются степень инфильтрации кожи, наличие/отсутствие кожных узлов, выраженность лимфаденопатии, степень тяжести поражения крови, уровень повышения ЛДГ и лейкоцитов в периферической крови), скорости его прогрессирования и влияния на качество жизни пациента. При назначении терапии рекомендовано соблюдать следующие принципы:
- по возможности избегать подавления иммунного ответа, назначение иммуномодулирующей терапии является предпочтительным;
- проведение комбинированной или мультимодальной (например, сочетание системной иммуномодулирующей и наружной) терапии приводит к более эффективным результатам, чем проведение любой монотерапии;
- своевременная диагностика и лечение инфекционных осложнений (иногда даже при отсутствии клинических признаков инфекционного процесса на коже) приводит к улучшению состояния пациента;
- большое значение имеет лечение зуда, значительно снижающего качество жизни.
Из-за выраженной гетерогенности и низкой распространенности заболевания количество контролируемых клинических исследований невелико, поэтому все рекомендации данного раздела имеют уровень доказательности C-D.
Схемы лечения
Виды комбинированной терапии, используемые при лечении синдрома Сезари (терапия первой линии)
| Комбинация | |
| системной терапии | наружной терапии |
| IFN-α | ПУВА |
| Метотрексат | Топические глюкокортикостероидные препараты |
| ЭКФ | ТОК |
| IFN-α | ТОК |
| Комбинация | |
| системной терапии | системной терапии |
| ЭКФ | IFN-α |
| ЭКФ | Метотрексат |
| IFN-α | Метотрексат |
ПУВА - псорален+ультрафиолетовое облучение спектра А
ЭКФ – экстракорпоральный фотоферез
ТОК – тотальное облучение кожи
Адьювантная терапия.
Наружные и системные глюкокортикостероидные препараты (10-20 мг преднизолона в сутки) используются в виде поддерживающей терапии у пациентов с СС. При длительном применении их отмена обычно ассоциирована с рецидивом заболевания, побочные эффекты включают атрофию кожи (при длительном наружном применении) и подавление функции надпочечников и/или остеопороз (при распространенной аппликации наружных или длительном приеме системных глюкокортикостероидных препаратов).
К дополнительным видам терапии относится фототерапия: ПУВА-терапия и узковолновое УФО спектра В (311 нм) (см. КР «Грибовидный микоз»).
Применение лейкафереза улучшает результаты стандартных видов терапии, уменьшает зуд и количество клеток Сезари в крови.
Тотальное облучение кожи (ТОК) в дозе 20-40 Гр рекомендовано комбинировать с другими видами системной терапии или в виде монотерапии с паллиативными целями.
Большое значение в ведении пациентов с СС имеет терапия, направленная на снижение интенсивности зуда и различных нейропатий (ощущений жжения, боли, стягивания кожи, парестезий). Для уменьшения этих ощущений используются увлажняющие кремы и антигистаминные препараты. Известно, что кожа больных СС избыточно колонизирована S.aureus, поэтому антибиотикотерапия приводит не только к снижению зуда, но и к улучшению течения заболевания. При выраженном зуде рекомендовано назначение габапентина – препарата, используемого для лечения нейропатических болей. Начинают с дозы 900 мг в сутки в 3 приема и постепенно увеличивают дозу до 3600 мг в сутки. Побочный эффект в виде седации позволяет пациентам нормализовать ночной сон. Для усиления снотворного эффекта в ночное время к терапии можно применять 7,5-15 мг миртазапина на ночь.
Критерии эффективности лечения
При СС используются критерии ответа на лечение, предложенные ISCL, EORTC и Американским консорциумом по кожным лимфомам (USCLC) (см. КР «Грибовидный микоз»).
Информация
Источники и литература
Информация
Персональный состав рабочей группы по подготовке федеральных клинических рекомендаций по профилю "Дерматовенерология", раздел «Лимфомы кожи»:
1. Белоусова Ирена Эдуардовна – доцент кафедры кожных и венерических болезней Военно-медицинской академии им. С. М. Кирова, доктор медицинских наук, г. Санкт-Петербург.
2. Самцов Алексей Викторович - заведующий кафедрой кожных и венерических болезней Военно-медицинской академии им. С. М. Кирова, доктор медицинских наук, профессор, г. Санкт-Петербург.
Эксперты-патоморфологи:
Байков В.В., Ковригина А.М., Криволапов Ю.А., Мационис А.Э., Петров С.В.
Эксперты-радиологи:
Ильин Н.В., Сотников В.М., Трофимова О.П.
МЕТОДОЛОГИЯ
Методы, использованные для сбора/селекции доказательств:
поиск в электронных базах данных.
Описание методов, использованных для сбора/селекции доказательств:
доказательной базой для рекомендаций являются публикации, вошедшие в Кокрановскую библиотеку, базы данных EMBASE и MEDLINE.
Методы, использованные для оценки качества и силы доказательств:
· Консенсус экспертов;
· Оценка значимости в соответствии с рейтинговой схемой (схема прилагается).
Рейтинговая схема для оценки силы рекомендаций:
| Уровни доказательств | Описание |
| 1++ | Мета-анализы высокого качества, систематические обзоры рандомизированных контролируемых исследований (РКИ) или РКИ с очень низким риском систематических ошибок |
| 1+ | Качественно проведенные мета-анализы, систематические, или РКИ с низким риском систематических ошибок |
| 1- | Мета-анализы, систематические, или РКИ с высоким риском систематических ошибок |
| 2++ | Высококачественные систематические обзоры исследований случай-контроль или когортных исследований. Высококачественные обзоры исследований случай-контроль или когортных исследований с очень низким риском эффектов смешивания или систематических ошибок и средней вероятностью причинной взаимосвязи |
| 2+ | Хорошо проведенные исследования случай-контроль или когортные исследования со средним риском эффектов смешивания или систематических ошибок и средней вероятностью причинной взаимосвязи |
| 2- | Исследования случай-контроль или когортные исследования с высоким риском эффектов смешивания или систематических ошибок и средней вероятностью причинной взаимосвязи |
| 3 | Неаналитические исследования (например: описания случаев, серий случаев) |
| 4 | Мнение экспертов |
Методы, использованные для анализа доказательств:
· Обзоры опубликованных мета-анализов;
· Систематические обзоры с таблицами доказательств.
Методы, использованные для формулирования рекомендаций:
Консенсус экспертов.
Рейтинговая схема для оценки силы рекомендаций:
| Сила | Описание |
| А | По меньшей мере один мета-анализ, систематический обзор или РКИ, оцененные как 1++ , напрямую применимые к целевой популяции и демонстрирующие устойчивость результатов или группа доказательств, включающая результаты исследований, оцененные как 1+, напрямую применимые к целевой популяции и демонстрирующие общую устойчивость результатов |
| В | Группа доказательств, включающая результаты исследований, оцененные как 2++, напрямую применимые к целевой популяции и демонстрирующие общую устойчивость результатов или экстраполированные доказательства из исследований, оцененных как 1++ или 1+ |
| С | Группа доказательств, включающая результаты исследований, оцененные как 2+, напрямую применимые к целевой популяции и демонстрирующие общую устойчивость результатов; или экстраполированные доказательства из исследований, оцененных как 2++ |
| D | Доказательства уровня 3 или 4; или экстраполированные доказательства из исследований, оцененных как 2+ |
Индикаторы доброкачественной практики (Good Practice Points – GPPs):
Рекомендуемая доброкачественная практика базируется на клиническом опыте членов рабочей группы по разработке рекомендаций.
Экономический анализ:
Анализ стоимости не проводился и публикации по фармакоэкономике не анализировались.
Метод валидизации рекомендаций:
· Внешняя экспертная оценка;
· Внутренняя экспертная оценка.
Консультация и экспертная оценка:
Предварительная версия была выставлена для обсуждения на сайте ФГБУ «Государственный научный центр дерматовенерологии и косметологии» Минздрава России для того, чтобы лица, не участвующие в разработке рекомендаций, имели возможность принять участие в обсуждении и совершенствовании рекомендаций.
Рабочая группа:
Для окончательной редакции и контроля качества рекомендации повторно проанализированы членами рабочей группы.
Основные рекомендации:
Сила рекомендаций (A–D) приводится при изложении текста рекомендаций.
Аутоиммунный лимфопролиферативный синдром – группа генетически обусловленных заболеваний, которые возникают по причине наследственных или соматических мутаций в генах, отвечающих за различные этапы FAS-обусловленного апоптоза. Симптоматика может быть вариабельной и наиболее часто включает в себя лимфаденопатию, спленомегалию и разнообразные аутоиммунные поражения системы крови, печени, щитовидной железы. Диагностика аутоиммунного лимфопролиферативного синдрома производится на основании результатов общего и биохимического анализов крови, биопсии лимфатических узлов, генетических исследований. Специфического лечения заболевания в настоящий момент нет, применяют комбинации иммунносупрессивной и цитотоксической терапии.
Общие сведения
Данные о встречаемости у разных исследователей довольно сильно различаются, на сегодняшний момент описано более 500 случаев различных форм аутоиммунного лимфопролиферативного синдрома. Наследственные формы заболевания передаются по аутосомно-доминантному типу, при этом в развитии врожденных форм также довольно велика роль спонтанных мутаций. Среди больных с одинаковой частотой встречаются как мальчики, так и девочки.
Причины
Выяснено, что причиной любого типа АЛС является нарушение FAS-опосредованного апоптоза лимфоцитов. При образовании Т-лимфоцитов те линии, которые способны атаковать собственные ткани, уничтожаются за счет активизации рецепторов CD-95 (Fas-рецепторов) на поверхности их мембраны. Активация CD-95, относящегося к группе рецепторов фактора некроза опухолей, запускает многостадийную реакцию с участием каспаз, которая оканчивается апоптозом клетки.
При аутоиммунном лимфопролиферативном синдроме генетические мутации приводят к блоку этого процесса на определенном этапе, из-за чего устранения потенциально опасных клонов Т-лимфоцитов не происходит, и они начинают накапливаться в лимфатических узлах. Кроме того, создаются условия для аутоиммунного поражения органов и тканей.
Наиболее часто встречаются наследственные и спонтанные мутации в гене TNFRSF6, который кодирует собственно Fas-рецептор. При этом нарушение структуры белка (особенно домена, отвечающего за взаимодействие с FADD-молекулой) приводит к тому, что он становится неспособным выполнять свои рецепторные функции и активизировать апоптоз. Возможны и соматические мутации в гене FAS, которые в полной мере проявляют себя в позднем детском или подростковом периоде, и поэтому их относят к отдельной группе АЛС.
Второй по распространенности вариант аутоиммунного лимфопролиферативного синдрома обусловлен мутацией в гене CASP10, кодирующем цистин-аспарагин кислотную протеазу (каспаза-10). Этот белок играет ключевую роль в передаче сигнала об апоптозе с клеточной мембраны в ядро клетки. К этому же варианту относят и мутации гена CASP8.
Третьим по распространенности является аутоиммунный лимфопролиферативный синдром, который вызван мутацией в гене FASLG, кодирующем Fas-лиганд или рецептор CD-178. Он играет вспомогательную роль в распознавании факторов, стимулирующих апоптоз, и участвует в передаче сигнала в клетку.
Некоторые формы АЛС обусловлены мутацией гена NRAS, который кодирует «малый G-белок», принимающий участие в качестве вторичного мессенджера в передаче сигналов с мембраны в клетку, в том числе и ядро. Примерно в трети случаев аутоиммунного лимфопролиферативного синдрома врачам-иммунологам не удается установить непосредственную причину заболевания.
Классификация аутоиммунного лимфопролиферативного синдрома
При помощи методов современной генетики удалось выявить шесть основных форм АЛС:
ALPS 1A – вызвана мутацией гена TNFRSF6, расположенного на 10-й хромосоме, чаще всего имеет врожденный характер, наследуется по аутосомно-доминантному типу. По статистике, более 40% АЛС относятся именно к этой разновидности.
ALPS 1В – обусловлена мутацией гена FASLG, также довольно часто приводит к врожденному аутоиммунному лимфопролиферативному синдрому. К этому типу относят около 10% от всех клинических случаев АЛС.
ALPS 1m – ее причиной являются соматические мутации в гене FAS, возникающие в детском или подростковом возрасте и поэтому приводящие к поздним формам АЛС. При этом повреждение гена должно произойти в полипотентной клетке-предшественнице, которая способна дать начало многим линиям лимфоцитов. При этой форме наиболее часто возникает внезапная самопроизвольная ремиссия заболевания.
ALPS 2 – вызвана мутацией в генах CASP10 и, по некоторым данным, CASP8, которые кодируют белки-каспазы, передающие сигнал об апоптозе от рецептора к ядру клетки. Эта форма аутоиммунного лифопролиферативного синдрома составляет примерно 25% от всех случаев заболевания, может быть как врожденной, так и проявиться в более старшем возрасте.
ALPS 3 – мутация какого гена и характер ее наследования при этой форме неизвестны. Особенностью такого варианта АЛС является нарушение не только FAS-, но и IL2-опосредованного апоптоза, а также более тяжелый характер течения.
ALPS 4 – обусловлена мутацией гена NRAS, также кодирующего белки-передатчики внутриклеточного сигнала. Данный тип аутоиммунного лимфопролиферативного синдрома характеризуется более доброкачественным течением и умеренной выраженностью симптомов.
Симптомы аутоиммунного лимфопролиферативного синдрома
Симптомы АЛС довольно вариабельны из-за большого количества мутаций, которые могут приводить к такому состоянию. Начало заболевания можно заметить уже на 15-й день после рождения (при врожденных формах), в детском или подростковом возрасте в случае соматических мутаций в генах FAS, CASP10 или NRAS. Обычно первым проявлением заболевания является лимфаденопатия – подмышечные, паховые или шейные лимфатические узлы увеличиваются в размерах, но при этом безболезненны и не спаяны с окружающими тканями. Регистрируется спленомегалия, в некоторых случаях она сопровождается увеличением печени (гепатоспленомегалия).
Аутоиммунные проявления АЛС регистрируются обычно через некоторое время после лимфаденопатии и увеличения селезенки. В основном это поражения кровяных ростков – тромбоцитопения, гемолитическая анемия, приводящая к желтухе, изредка нейтропения. Помимо крови, аутоиммунному поражению могут подвергаться органы ЖКТ (возникают гастрит, панкреатит, колит, аутоиммунный гепатит). На коже могут проявляться признаки васкулита, делая клинику аутоиммунного лимфопролиферативного синдрома схожей с таковой при системной красной волчанке. Кроме того, могут возникать аутоиммунные формы тиреоидита, гломерулонефрита, поражаться суставы, ткани глаза (иридоциклит, увеит). Нередки поражения центральной нервной системы – эпилептические припадки, миелиты, мозжечковая атаксия.
Выраженность симптомов и их количество может значительно варьироваться у каждого конкретного больного. Кроме того, при аутоиммунном лимфопролиферативном синдроме в десятки раз возрастает риск развития злокачественных опухолей, так как опухолевые клоны лимфоцитов также устраняются посредством апоптоза.
Примерно в 20% случаев АЛС приводит к неходжкинским лимфомам (лимфома Беркитта, фолликулярная лимфома), описаны и другие онкологические заболевания. Из-за этого проявления АЛС могут быть ошибочно определены как следствие опухолевой инфильтрации лимфоидной ткани. Среди других осложнений аутоиммунного лимфопролиферативного синдрома наиболее часто встречается травматический разрыв селезенки, сепсис и другие инфекционные поражения.
Диагностика
Диагностика АЛС производится на основании осмотра, а также лабораторных, иммунологических и генетических исследований. При осмотре выявляют увеличение более чем трех групп лимфатических узлов, спленомегалию, увеличение печени. Анализ крови может показывать уменьшение количества некоторых клеток (анемию, тромбоцитопению), у части больных определяется высокая (до 30%) эозинофилия. Проба Кумбса положительная, в биохимическом анализе крови определяется выраженная гипергаммаглобулинемия.
Врачом-генетиком может быть произведено секвенирование гена FAS с целью выявления мутаций, ставших причиной аутоиммунного лимфопролиферативного синдрома. С учетом значительной величины этого гена для ускорения и удешевления процедуры поиск может быть произведен лишь в отдельных экзонах гена FAS, в которых наиболее часто обнаруживаются нарушения – эти участки называют «горячими точками». Таким образом, при помощи генетической диагностики можно определить АЛС только 1А, 1В и 1m типов. Методики определения остальных форм АЛС генетическими методами на сегодняшний день не разработаны. Изучение наследственного анамнеза в ряде случаев будет неэффективно из-за значительной доли форм заболевания, вызванных соматическими мутациями.
Лечение аутоиммунного лимфопролиферативного синдрома
Этиотропное лечение аутоиммунного лимфопролиферативного синдрома не разработано, патогенетическая терапия сводится к применению иммуносупрессивных и цитотоксических средств. В качестве средств, подавляющих аутоиммунную активность, наиболее часто используют кортикостероиды (преднизолон, дексаметазон). К специфическим препаратам, ограничивающим скорость пролиферации лимфоцитов, относят микофенолата мофетил, сиролимус. Также при аутоиммунном лимфопролиферативном синдроме активно применяются традиционные цитотоксические средства – метотрексат, циклоспорин А и другие.
При значительном увеличении селезенки или отсутствии эффекта от консервативного лечения прибегают к спленэктомии. Пересадка костного мозга и использование стволовых клеток в долгосрочной перспективе давали только временный эффект. При значительно выраженных гематологических нарушениях применяют гемотрансфузии, введение эритроцитарной или тромбоцитарной массы. Больному следует избегать физических нагрузок, использовать высоковитаминную диету.
Прогноз
Прогноз заболевания, ввиду высокой вариабельности и выраженности симптомов, неопределенный или неблагоприятный. У большей части больных проявления заболевания постепенно нарастают, со временем приводя к летальной анемии, тромбоцитопении, билиарному циррозу печени. Также важную роль в прогнозе играют нарушения иммунитета, так как нередко причиной смерти выступают сепсис и другие инфекционные поражения. В прогнозе аутоиммунного лимфопролиферативного синдрома следует учитывать и повышенный риск онкологических заболеваний, примерно пятая часть больных умирает от различных типов лимфом. В некоторых случаях возникает спонтанная и длительная ремиссия патологии.
Псевдолимфомы кожи – группа доброкачественных реактивных дерматозов, характеризующихся гиперплазией лимфоидной ткани. Особенностью патологического процесса является локализация первичных элементов (эритем, узелков, бляшек) на лице, при некоторых видах псевдолимфомы может наблюдаться диссеминация высыпаний. Постановка диагноза осуществляется клинически с учётом анамнеза патологии (провоцирующих факторов), результатов гистологии, ПЦР-диагностики и иммуногистохимического тестирования. Лечение включает детоксикационную терапию, антигистаминные средства, кортикостероиды, энтеросорбенты и физиотерапию.
Общие сведения
Псевдолимфомы кожи – совокупность пролиферативных процессов лимфоидной ткани доброкачественного характера, способных к разрешению после курса базовой терапии или удаления провоцирующего агента. Патологический процесс встречается на всех географических широтах, не имеет расовых и сезонных различий. Впервые термин псевдолимфома был введён в дерматологическую практику в 1891 году М. Капоши. В 70-х годах прошлого столетия на основании иммуногистохимических методов исследования псевдолимфомы удалось разделить на две группы: Т-клеточные и В-клеточные – в зависимости от типа доминирующих в лимфоидном инфильтрате клеток. В-клеточные псевдолимфомы кожи чаще диагностируются у детей и подростков, Т-клеточные – у взрослых мужчин.
Современные дерматологи признают термин псевдолимфомы условно. Многие практикующие врачи считают более верным предложенный Г. Вудом термин клональный дерматит, в основу которого положено выявление клона лимфоцитов, преобладающих в развитии патологического процесса. Актуальность проблемы обусловлена возможностью озлокачествления псевдолимфом, необходимостью их точной и своевременной диагностики из-за преимущественной локализации на лице, а также снижения качества жизни пациента из-за возникающего эстетического дефекта.
Причины псевдолимфом кожи
Причин развития псевдолимфом несколько, одни из них известны, как, например, укус иксодового клеща при В-клеточных псевдолимфомах, другие установлены предположительно, как например, инфекция, свет, аутоиммунные процессы и аллергия при Т-клеточных псевдолимфомах. Общим является реактивный ответ кожи в виде усиления лимфопролиферативных процессов.
Механизм развития патологии зависит от разновидности псевдолимфомы. В случае В-клеточных псевдолимфом активная пролиферация связана с изменением клеточного метаболизма. Важнейшим моментом является насыщение клетки кислородом. Для нормальной работы иммунной системы лимфоидным клеткам необходимо достаточное количество энергии, чтобы вырабатывать В-лимфоциты, стимулировать защитные механизмы и восстанавливать нарушенную целостность дермы. В случае псевдолимфомы повышенная потребность лимфоидной ткани во внутриклеточном кислороде не удовлетворяется, поскольку значительную часть кислорода получают В-лимфоциты, нейтрализующие антиген-триггер.
Кроме того, нормальное кислородоснабжение необходимо клеткам дермы, участвующим в восстановлении кожных покровов. В результате перераспределения кислорода у Т- и В-лимфоцитов, призванных вместе с клетками ретикулоэндотелиальной системы восстановить дефект кожи, наступает гипоксия, провоцирующая усиленное компенсаторное деление клеток. В процесс включаются фибробласты, клетки гистиоцитарного ряда и плазмоциты, которые при исследовании выявляются в лимфоцитарном инфильтрате.
В случае Т-клеточных псевдолимфом механизм развития лимфопролиферативных процессов несколько отличается, ведущая роль в нём отводится нарушению дифференцировки Т-лимфоцитов на фоне системных аутоиммунных заболеваний. Измененные Т-лимфоциты провоцируют усиленную пролиферацию клеток дермы, которые в результате антигенного повреждения начинают выработку биологически активных веществ, стимулирующих воспаление и активизирующих пролиферацию Т-клеток. В обоих случаях (как при Т-, так и при В-клеточных псевдолимфомах) образуется доброкачественный лимфопролиферативный инфильтрат.
Классификация псевдолимфом кожи
Единой классификации патологического процесса не существует. Рабочей классификацией в современной дерматологии принято считать разделение псевдолимфом по клеточному составу. Выделяют В-клеточные и Т-клеточные псевдолимфомы, состоящие преимущественно из В-лимфоцитов и Т-лимфоцитов.
Наиболее распространенной В-клеточной псевдолимфомой является доброкачественная лимфоплазия кожи (ДЛК, лимфоцитома, лимфаденоз доброкачественный Бефверстедта, псевдолимфома Шпиглера-Фендта), возникающая под влиянием экзогенных факторов. Первичным элементом является спонтанно регрессирующий и вновь появляющийся бурый тестообразный узелок с чёткими границами, локализующийся на лице, ушных раковинах и в подмышечных впадинах. Различают:
- Постборрелиозную разновидность В-клеточной ДЛК – формируется во второй стадии боррелиоза, проявляется папулами и бляшками.
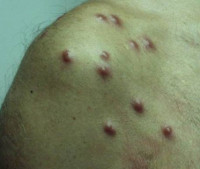
- Постскабиозную разновидность В-клеточной ДЛК (персистирующая узловая реакция на укусы насекомых, постскабиозная лимфоплазия кожи) – персистирующую псевдолимфому, зудящий дерматоз, первичным элементом которого являются папулы, локализующиеся в области половых органов, в подмышечных впадинах, на ягодицах и локтях.
Выделяют следующие разновидности Т-клеточных псевдолимфом:
- Лимфоцитарная инфильтрация Джесснера-Канофа – хронический доброкачественный патологический процесс, первичным элементом которого является плоская розово-синюшная бляшка, локализующаяся на лице.
- Истинная псевдолимфома (ретикулярная гиперплазия, реактивный ретикулёз, синдром псевдолимфомы, эритродермия псевдолимфоматозная) – доброкачественный патологический процесс, возникающий в ответ на действие лекарственных средств, бензина, ароматических масел и некоторых пищевых продуктов. Псевдолимфома склонна к спонтанному разрешению, ее отличительная черта – способность быть маркером латентного опухолевого процесса в организме. Клинически всегда сопровождается продромальными явлениями, первичным элементом является сливная эритема.
- Лимфоматоидный папулёз – доброкачественная медленно растущая узелковая опухоль кожи, первичным элементом является бурое пятно без излюбленной локализации.
- Актинический ретикулоид – редкий хронический фотодерматоз, гистологически напоминающий грибовидный микоз. Первичным элементом является эритема, возникающая под действием световых лучей и трансформирующаяся в эритродермию.
Симптомы псевдолимфом кожи
Клинические проявления Т-клеточных и В-клеточных псевдолимфом разнятся. В-клеточные псевдолимфомы чаще поражают детей и подростков, сопровождаются реактивным высыпанием мелкоузелковых или бляшечных розовато-фиолетовых элементов с четкими границами (одиночных или множественных), способных формировать более крупные инфильтраты. Локализация – лицо, шея, ушные раковины, подмышечные впадины, вокруг сосков молочной железы, на мошонке. Субъективное состояние пациентов не нарушено, в процесс часто вовлекаются регионарные лимфоузлы.
Т-клеточные псевдолимфомы встречаются у взрослых мужчин в виде спонтанного высыпания на лице, туловище и конечностях. Представляют собой синюшные папулы, растущие по периферии, образующие очаги инфильтративного поражения с регрессом в центральной части и самостоятельным разрешением. С течением времени процесс возобновляется на тех же участках кожного покрова или на неповреждённой коже.
Диагностика и лечение псевдолимфом кожи
Диагностика псевдолимфомы с неустановленной причиной возникновения представляет собой достаточно сложную задачу. Необходимо исключить злокачественный процесс, поэтому наряду с клиническими и анамнестическими данными используют результаты гистологического исследования. Обязательно проводят ПЦР-диагностику, иммуногистохимическое тестирование (выражена экспрессия иммуноглобулиновых лёгких цепей лямбда и каппа) и Саузерн-блот-анализ, позволяющий выявлять дефектные гены.
В-клеточные псевдолимфомы дифференцируют с иммуноцитомой, лимфосаркомой, В-лимфомой, лимфомой MALT-типа, саркоидозом, туберкулёзной волчанкой и эозинофильной гранулёмой лица. Дифференциальную диагностику Т-клеточных псевдолимфом проводят с токсикодермией, красной волчанкой, другими вариантами псевдолимфом, лимфомами кожи, каплевидным псориазом, папулонекротическим ангиитом, папулонекротическим туберкулёзом, грибовидным микозом, синдромом Сезари, фотодерматозами и узловатой почесухой.
Терапию осуществляет дерматолог, при необходимости к лечению подключают онколога и косметолога. Приоритет отдают детоксикационной терапии (гемодез, плазмаферез, гемосорбция). Используют кортикостероиды внутрь и наружно, назначают энтеросорбенты, антигистаминные препараты, аминохинолины, цитостатики и нестероидные противовоспалительные средства. В локальные очаги вводят глюкокортикоиды путем инъекций. Применяют ПУВА-терапию. Прогноз относительно благоприятный, необходимо регулярное наблюдение у дерматолога.
Лимфома кожи — опухолевые поражения кожи, возникающие в результате злокачественного размножения в ней лимфоцитов. В зависимости от вида размножающихся лимфоцитов различают Т- и В-клеточные лимфомы. Заболевание проявляется образованием на коже узелков, бляшек или эритродермических участков, что сопровождается увеличением лимфатических узлов. Диагностика проводится путем гистологического исследования биопсийного материала из пораженного участка. В лечении лимфомы кожи применяется химиотерапия, лучевая терапия, ПУВА-терапия, экстракорпорального фотофорез.
Общие сведения
По данным исследований Т-клеточные лимфомы кожи встречаются в 65-70% случаев, тогда как В-клеточные лимфомы кожи составляют 20-25%. Еще 10% занимают так называемые неклассифицируемые лимфомы кожи.
Причины возникновения лимфомы кожи
Развитие лимфомы кожи связано с мутацией Т или В лимфоцитов, которая приводит к их бесконтрольному размножению и миграции в кожу. Точные причины, запускающие этот механизм не известны. Предполагают, что возникновение злокачественного клона лимфоцитов может быть спровоцировано постоянной антигенной стимуляцией на фоне нарушенной иммунной защиты организма.
Провоцирующую роль отводят вирусным инфекциям, вызванным ретровирусами, цитомегаловирусом, вирусом простого герпеса 8 типа, вирусом Эпштейна—Барра. Действие различных химических веществ и канцерогенов, применяющихся в сельском хозяйстве, химической промышленности, строительстве и других областях, также может быть причиной возникновения лимфомы кожи.
Лимфома кожи бывает первичной, когда заболевание начинается с поражения дермы, и вторичной — в результате миграции лимфоцитов из лимфоидного органа, в котором происходит их размножение. К таким органом относится костный мозг, вилочковая железа, лимфатические узлы, селезенка, лимфоидные скопления по ходу дыхательных путей и желудочно-кишечного тракта.
Симптомы лимфомы кожи
Лимфомы кожи характеризуются полиморфизмом сыпи (пятна, бляшки, узлы), различной степенью выраженности зуда и увеличением периферических лимфатических узлов. По степени злокачественности выделяют лимфомы I, II и III степени. По клиническим проявлениям: узелковую, бляшечную и эритродермическую формы. Узелковая форма Т-клеточной лимфомы кожи I степени характеризуется мелкими плоскими узелками размером с просяное зерно. Узелки имеют сиреневый или желтоватый цвет, располагаются группами и склонны к спонтанному регрессу. При более злокачественном течении узелки увеличиваются, приобретают вишневый цвет и утрачивают тенденцию к группировке. Пациенты погибают от метастазов через 2-5 лет.
Редко встречается мелкоузелковая форма Т-клеточной лимфомы кожи, при которой фолликулярные узелки сливаются в бляшки с напоминающим псориаз поверхностным шелушением. На этом фоне появляются крупные узелки, которые затем подвергаются некрозу. Бляшечная форма Т-клеточной лимфомы кожи I степени представлена нерезко отграниченными бляшками желтоватого цвета. Размер бляшек может быть больше ладони. Они постепенно разрешаются с образованием участков атрофии и гиперпигментации.
Бляшечная форма II степени (грибовидный микоз Алибера) встречается в 26% всех лимфом кожи. Для нее характерно стадийное развитие. Вначале появляются шелушащиеся ярко-розовые пятна и другие элементы (эритематозная стадия). Затем на месте пятен формируются застойно-красные бляшки часто с мокнущей поверхностью и периферическим ростом (бляшечная стадия). В опухолевой стадии бляшки сменяются плоскими узлами размером до апельсина с некрозом в центре образования.
Эритродермическая форма Т-клеточной лимфомы кожи I степени (синдром пре-Сезари) часто развивается на фоне длительно, в течение 10-15 лет, существующей экземы или нейродермита. Кожа покрасневшая и отечная, покрыта крупными пластинками белых чешуек. Наблюдается генерализованное увеличение лимфоузлов, дистрофия ногтей, выпадение волос, лихорадка и мучительный зуд. Через несколько лет пациент может погибнуть от кахексии или процесс переходит в эритродермическую форму II степени (синдром Сезари), характеризующуюся выраженной инфильтрацией, шелушением и сухостью кожи.
Для В-клеточных лимфом кожи характерно отсутствие зуда и других субъективных ощущений при I и II степени злокачественности. Они проявляются бляшечной и узловатой формой. Для бляшечной формы характерны те же стадии, что и для Т-клеточной лимфомы кожи. Узловатая форма развивается с образованием одного или нескольких полушаровидных узлов плотно-эластической консистенции, величина которых достигает размеров грецкого ореха.
Диагностика лимфомы кожи
Во многих случаях лимфомы кожи сопровождаются изменениями в клиническом анализе крови. Для Т-клеточной лимфомы характерны лейкопения и моноцитоз. При синдроме пре-Сезари наблюдается лейкоцитоз и нейтрофилез, повышение количества эозинофилов. При синдроме Сезари может наблюдаться увеличение лейкоцитов до 30000-200000. В-клеточные лимфомы кожи характеризуются возникновением вначале нормохромной, а затем гемолитической анемии.
Решающее диагностическое значение имеет гистологическое и цитологическое исследование материала, взятого путем биопсии элементов лимфомы кожи, а при необходимости и лимфатических узлов. Биопсия позволяет дифференцировать Т и В клеточную лимфому кожи, а также определить степень ее злокачественности. При вовлечении в процесс внутренних органов проводят их исследование: УЗИ брюшной полости, рентгенографию легких, КТ легких и т. п.
Лечение и прогноз лимфомы кожи
Основным методом лечения пациентов с лимфомой кожи является химиотерапия. В ней используют цитостатики (винкристин, винбластин, циклофосфан), кортикостероиды (преднизолон, бетаметазон) и интерфероны (гамма-интерферон). В лечении отдельных пятен, бляшек и единичных опухолей применяют лучевую терапию, ПУВА-терапию, фототерапию. В некоторых случаях эффективно проведение экстракорпорального фотофореза. Часто комбинируют различные методы лечения и применяемые препараты. Например, облучение назначается совместно с химиотерапией и после нее.
При своевременном начале лечения и I-II степени злокачественности лимфомы кожи часто удается добиться выраженной ремиссии и продлить жизнь пациента. К летальному исходу в таком случае приводят интеркуррентные заболевания или осложнения лечения. Если лимфома кожи диагностирована в опухолевой стадии или имеет выраженную злокачественность, прогноз крайне неблагоприятный, летальный исход может наступить через 2 года после начала заболевания.
Читайте также: