
Что такое генетическое заболевание кожи у детей
Обновлено: 25.04.2024
Мышечная дистрофия Дюшенна (МДД) – редкое генетическое заболевание, которое обычно поражает мальчиков (примерно одного из 3500–5000). Болезнь развивается из-за мутаций в гене дистрофина и разрушает мышцы: сначала ребенок перестает ходить, а со временем и дышать. Пока заболевание остается неизлечимым.
Эту болезнь врачи в России часто диагностируют поздно. Ольга Гремякова, мама семилетнего Гордея, сама догадалась, что у ее сына миодистрофия Дюшенна: заметила симптомы, нашла информацию и отвезла сына к врачам, которые подтвердили ее опасения.
Ольга и ее семья создали благотворительный фонд «Гордей», чтобы улучшать качество медицинской, социальной и психологической помощи пациентам с МДД и их семьям. В декабре фонд провел первую в России конференцию для родителей и врачей, посвященную миодистрофии Дюшенна.
Поговорили с Ольгой о том, как распознать болезнь и с чем сталкиваются пациенты с МДД и их семьи после постановки диагноза.
От первых симптомов до диагноза
Прислушиваться к себе
Я видела, что Гордей отличается от других ребят, но не понимала, почему. До трех лет он не разговаривал. Был немного неуклюжий, не умел прыгать и бегать. Мне говорили, что причина в тяжелых родах, поэтому нужно немного подождать, и он перерастет эту неуклюжесть. Либо мягко намекали на то, что я просто тревожная мама, которая беспокоится за своего первого малыша. Когда мы проходили обследования перед детским садом, все специалисты в очередной раз сказали, что сын здоров. Не насторожился никто – ни врачи, ни моя мама, доктор медицинских наук.
Кроме того, Гордей был и остается очень требовательным и чувствительным ребенком, с ним сложно взаимодействовать. Родные говорили, что это я воспитала его таким: он капризный и просится на ручки не из-за усталости, а потому что манипулирует. Ну что скажешь в этот момент? Я действительно думала, что сама виновата. Тогда я не знала, что эти особенности тоже связаны с его болезнью.
Заметить симптомы
Я услышала о миодистрофии Дюшенна за пару лет до того, как ее диагностировали у Гордея. Это была история Ольги и Петра Свешниковых – учредителей фонда «Мой Мио», которые усыновили ребенка с МДД. Этот поступок вызвал у меня бесконечные уважение и восхищение. Но тогда я просто прочла об этом и продолжила жить дальше. Спустя какое-то время я увидела в фейсбуке историю папы и сына с МДД. После фразы «я смотрел на его икры, они были такие мускулистые, а сил-то в них не было» картинка в голове полностью сложилась. Я погуглила симптомы и поняла, что с моим ребенком.
Главный признак мышечной дистрофии – прогрессирующая мышечная слабость. Специфические симптомы появляются в разном возрасте и в разных группах мышц, в зависимости от типа мышечной дистрофии. Обычно первые признаки МДД проявляются к двум-трем годам. Ребенок может часто падать, медленно бегать, ходить на цыпочках или вразвалочку. Ему может быть сложно вставать с пола и подниматься по лестнице. Часто у ребенка с МДД непропорционально большие икры. Сначала слабость более выражена в мышцах бедер, но со временем она распространяется и на другие, включая мышцы, участвующие в дыхании. У ребенка может быть задержка двигательного и речевого развития, трудности в обучении. МДД также влияет на память, манеру общения и эмоциональное состояние.
Уже через пару часов Гордей сдал кровь. Как мы дожили до конца дня, я не помню. Ночью проснулась, проверила почту и нашла письмо от лаборатории: уровень креатинкиназы – 23076 при норме у мальчиков до 200!
На следующий вечер я укладывала сына спать и сказала ему: «Гордей, я теперь все про тебя знаю. Знаю, что ты устаешь, что у тебя не очень сильные мышцы, что ты такой вот особенный малыш». Гордюше тогда было четыре с половиной года. Он ничего не сказал, а просто тихонечко заплакал. Наверное, для него было важно, что его наконец понимают.
При подозрении на МДД берут анализ крови на креатинкиназу (КК) – фермент, который мышцы выделяют при повреждении. Высокий уровень КК – характерный признак миодистрофии Дюшенна. Для подтверждения диагноза используют генетические тесты – они находят крупные мутации в гене дистрофина. Если крупных мутаций не нашли, врачи сделают секвенирование (расшифровку последовательности) гена, чтобы найти мелкие точечные мутации.
Если обращать внимание на характерные симптомы, то диагностировать МДД можно рано – например, с года до двух. Но для этого врачи должны заметить признаки миодистрофии Дюшенна в потоке пациентов. По мнению Ольги, идеальным диагностическим решением было бы добавить к неонатальному скринингу тест на креатинкиназу – он простой и стоит около 400 рублей.
К сожалению, примерно половина мальчиков с МДД исходно получает неверный диагноз. Чаще всего гепатит, потому что из-за распада мышц растет уровень ферментов трансаминаз, что обычно соотносят с болезнью печени. Также мальчикам ставят перинатальное поражение ЦНС, аутизм, задержки двигательного и речевого развития и даже ДЦП. Иногда МДД диагностируют очень поздно – в возрасте от 8 до 12 лет, когда симптоматика становится заметной и папы начинают носить мальчиков на руках.
 |
Ольга с детьми. Фото из личного архива
Делать все возможное
Первое изменение в жизни моей семьи – это зашкаливающий уровень боли. Боли от диагноза нашего любимого малыша, которого еще пару дней назад мы считали здоровым. Вскоре у меня развилось состояние предвосхищающего горя: ты знаешь, что ребенок жив, но думая о его диагнозе, понимаешь – все предопределено. Рухнуло все: представление о счастливом материнстве, где я – мама двух прекрасных здоровых малышей, о будущем, где сын будет катать меня на машине и мы вместе будем делать всякие штуки. Рухнула моя идентичность – как мамы и как женщины.
Я плохо помню первые два года жизни после постановки диагноза. Через полгода у меня началась депрессия: не было сил даже на самые обычные дела, я чувствовала себя разбитой, ничто меня не радовало… Мое состояние заметила мама и настояла на обращении к врачу – сама я, несмотря на психологическое образование, депрессии у себя не заметила. Врачи помогли. Все же я не могла пару месяцев лежать лицом к стене – нужно было заниматься детьми.
Мы решили, что будем пользоваться каждым днем и делать для Гордея все возможное, чтобы он чувствовал себя лучше. Да, волшебной таблетки у нас нет, но мы в силах снизить риск контрактур (ограничение подвижности сустава – прим. авт.) голеностопа и других осложнений, которые усаживают мальчиков в коляску раньше.
Физические упражнения, растяжка и низкоинтенсивные аэробные нагрузки (ходьба и плавание) помогают пациентам с МДД сохранить мышечную силу и диапазон движений в суставах. Специальные изделия – туторы – могут поддерживать голеностопный сустав и стопы в правильном положении, замедлять прогрессирование контрактур. Чем дольше ребенок ходит и чем позднее понадобится коляска, тем лучше для сердца и дыхательной системы и тем ниже риск развития сколиоза, требующего оперативного вмешательства.
Чтобы вернуть себе ресурс и сражаться за ребенка, с первого дня учитесь делать ему растяжки ахиллового сухожилия, голеней и бедер – по 20–30 минут ежедневно. Благодаря таким занятиям мальчик сможет ходить дольше. Когда родитель «своими руками» продлевает ребенку активные годы, это очень важно для обоих.
От непонимания до взаимной поддержки
Совершенствовать систему
Со временем мы узнали, как устроена помощь пациентам с миодистрофией Дюшенна. Помимо небольших выплат государство предлагает мальчикам санаторно-курортное лечение. Но пока система плохо понимает, что именно нужно детям с МДД. На опыте Гордея могу сказать, что предлагают обычно не то и не там – хоть и очень стараются. Два раза в год мальчикам и юношам с миодистрофией положена реабилитация – и это не массажи и грязи. Она конечно есть, но далеко не везде соответствует международным стандартам. Более того, не везде она человечная. Иногда мамы с сыновьями сбегают с реабилитации, потому что еда плохая, обращение еще хуже, занятий нет.
Также существует ИПРА – индивидуальный план реабилитации (абилитации). В него вписывают технические устройства, предлагаемые государством, например коляски и туторы. Но часто эти устройства не подходят ребенку, и родители вынуждены покупать другие за свой счет – при этом стоимость компенсируют не всегда. Это очень чувствительный вопрос для семьи, и самые большие трудности возникают именно со средствами передвижения для неходячих мальчиков.
Получается, что государство помогает, но сами механизмы негибкие и нуждаются в пересмотре. По международным стандартам реабилитации ребятам с МДД нужны теплый бассейн, правильные растяжки, полный медицинский чекап и сильная мультидисциплинарная команда. Еще год назад в России не было центров, где могут все это предложить. Сейчас самый современный вариант – реабилитация в Центральной клинической больнице, программу которой разработали эксперты ЦКБ в сотрудничестве с нашим фондом. Она соответствует лучшим международным практикам и в этом смысле уникальна для России.
Но такой центр один, а мальчики с МДД рождаются по всей стране. Их примерно четыре тысячи, при этом для системы здравоохранения видны не более полутора тысяч. В любом случае, наших мальчиков слишком много для того, чтобы всех отправлять в Москву. Ребятам нужна реабилитация в региональных центрах, ведь многим из них сложно передвигаться. Поэтому наша задача – создавать такие центры и обучать специалистов на местах. Для этого нам нужно сильное пациентское сообщество и некоммерческие организации, которые будут влиять на неповоротливую государственную систему и улучшать ее работу.
Отстаивать интересы
Мы стремимся к тому, чтобы в России приняли клинические рекомендации по ведению пациентов с МДД. Хотим, чтобы мальчики и их семьи получали все виды помощи – медицинскую, социальную и психологическую, и чтобы эта помощь соответствовала международным стандартам.
Есть и другие организации, занимающиеся миодистрофией Дюшенна – фонд «Мой Мио» (6 лет) и Родительский проект (3 года). У каждой из них более 500 подопечных. Они реализуют медицинские и образовательные программы для пациентов с МДД, оказывают информационную, консультативную и адресную помощь семьям.
После постановки диагноза семьи сталкиваются с дефицитом информации в отношении препаратов, физической терапии, пищевых добавок и приема стероидов. Именно потребность узнать больше помогает родителям разобраться в разных аспектах заболевания, а значит – принимать лучшие решения в интересах своего ребенка и сохранять чувство контроля.
Еще один способ вернуть контроль – присоединиться к родительскому сообществу. Помимо информации в нем можно найти опору, общаясь с другими родителями. У фонда есть сообщество «ProДюшенн», в котором более четырехсот участников, среди которых родители и врачи. Все наши семьи разные: у одних маленькие мальчики с МДД, у других юноши, а есть семьи, где сыновья уже ушли. Но цель у всех одна – поддерживать друг друга и отстаивать интересы пациентов с Дюшенном при взаимодействии с государством.
Также для нас важно, чтобы мамы мальчиков с МДД и их дочери могли выбирать свое репродуктивное будущее и знали об этом. Болезнь развивается из-за мутаций, которые в 70% случаев возникают в Х-хромосоме матери и передаются сыновьям. Если в семье уже есть мальчик с МДД, другие дети также могут унаследовать мутацию. Но предимплантационная диагностика и ЭКО позволяют планировать рождение здоровых детей после появления малыша с МДД. И если я решусь на третьего ребенка, то, скорее всего, обращусь к этим методам. Девочкам, чьи мамы – носительницы мутаций в гене дистрофина, можно сделать анализ ДНК и проверить, передалась ли мутация.
 |
Команда фонда «Гордей». Фото из личного архива
При МДД родители сталкиваются со множеством кризисных ситуаций. Часто травматичной становится сама постановка диагноза, когда результат генетического теста присылают по почте, а врачи отвечают на звонки без особого желания. Мальчик может столкнуться с переломом и потом долго восстанавливаться, либо теряет способность ходить. Он может оказаться в кресле и без перелома, когда происходит срыв компенсации – снижение мышечной силы. Молодому человеку может потребоваться респираторная поддержка, и это тоже может быть кризисным моментом – как и любое изменение в жизни, указывающее на переход болезни в более тяжелую стадию. И конечно, в таких ситуациях лучше работать с психологом индивидуально.
Еще есть переходный возраст. Это сложное время для любой семьи, но особенно для родителей мальчиков с МДД. Если ребенок принимает кортикостероиды, он не очень сильно растет – а расти ему важно. Для этого придется снизить дозу гормонов, но тогда он начнет слабеть. И этот момент пересмотра лечения, отношений с ребенком и его личных границ тоже бывает очень непростым.
У ребенка с МДД могут быть трудности с учебой, агрессивное поведение, тревожное расстройство или депрессия. Если знать об этом и вовремя обратить внимание, многие вещи можно скорректировать и улучшить качество жизни ребенка и родителей.
Недавно фонд запустил проект психологической поддержки родителей «Передышка». Мы провели уже три встречи, в начале следующего года надеемся параллельно запустить «Передышку» для пап, где будет психолог-мужчина, а также индивидуальную психологическую поддержку.
Искать лечение
Создать универсальное лекарство для МДД сложно. Во-первых, ген дистрофина огромный, и в нем может быть очень много разных мутаций – известно уже около 10 тысяч. Во-вторых, лекарства доставляют в клетки с помощью вирусных векторов (генетических конструкций – прим.авт.), у которых есть определенная емкость. Можно поместить в вирус только фрагмент гена дистрофина. Кроме того, у части пациентов имеется предсуществующий иммунитет к самим вирусам. В-третьих, чтобы лекарство проникло в мышцы, оно должно попасть в общий кровоток – а значит, пройти через печень, которая постарается удалить его из организма. Возникает конфликт: врачи пытаются доставить лекарство в мышцы, а тело сопротивляется и пытается его вывести.
Но таргетные препараты уже появились – и это большая подвижка. Со временем должно появится лекарство, которое подойдет многим пациентам с МДД. Значит нам нужно выявлять болезнь как можно раньше, чтобы быстрее начинать лечение.
24 ноября 2020 года в России зарегистрировали первый таргетный препарат для лечения МДД – аталурен. Он подходит 13% пациентов с МДД, имеющим нонсенс-мутации – точечные замены в гене дистрофина. По словам производителя, лекарство помогает восстанавливать уровень дистрофина и замедлять прогрессирование болезни. Существуют и другие препараты – вайондис, вилтепсо и экзондис, но они также предназначены для пациентов с определенными мутациями.
Традиционное лечение МДД направлено на устранение симптомов. Для повышения мышечной силы и продления двигательной активности врачи назначают пациенту кортикостероиды, но они увеличивают риск переломов и со временем могут привести к набору веса и повышению давления. Для сохранения функций сердечной мышцы применяют ингибиторы ангиотензинпревращающего фермента (АПФ) или бета-блокаторы.
Найти общий язык
Сейчас Гордей учится в нулевом классе. Он чувствует, что справляется, и совершенно счастлив. У него есть друзья, его хвалят учителя. Гордей учится играть в шахматы, с удовольствием изучает испанский и осваивает скетчинг. Для него и других ребят с МДД очень важны вопросы образования, социальной среды и общения.
Так же важна реакция врача на ребенка с миодистрофией. Если врач общается с ним тепло, уважительно и без страха – это одна история. Но когда врачи боятся диагноза, избегают общения с ребенком и ведут себя скованно – и родители, и сами дети это чувствуют. И то, как реагируют на ребенка врачи, учителя, чиновники на комиссиях по инвалидности – это вовсе не «сходил и забыл». Чувствуется негласная стигма «идите умирайте дома, лекарства нет» – и мы должны с ней бороться. Во-первых, лекарства появляются, их будет больше, они будут лучше. Во-вторых, в современном обществе никто не должен быть оставлен без помощи. Для этого нужно учиться делать все вместе – жить, работать, решать гражданские дела. И в этом смысле не может быть и речи о том, чтобы закрыться и угасать дома.
Сохранить веру
Нам всем нужна вера, и ее очень сложно сохранить. Особенно уязвимы родители взрослых сыновей. Ведь Дюшенн – одно из самых травмирующих заболеваний, причем травмируется вся семья, но особенно – мама, которая, как правило, осуществляет основной уход за сыном. Мамы сильно изранены, многим нужна психологическая поддержка и помощь в уходе за ребенком.
Бывает, что родители молодых людей говорят родителям мальчиков помладше: посмотрим, что вы скажете через 10 лет. Да, они испытали то, через что пока не прошла я. И тогда я думаю – действительно, а что я скажу через 10 лет? Я не знаю, я ли это буду или уже не я. Смогу ли сохранить себя, свою любовь, своего ребенка? Я делаю все, что могу, как и моя семья. Боюсь ли я будущего? Я надеюсь, что справлюсь. Ну а там – посмотрим, что я скажу через 10 лет.
Генодерматозы — это общее название неоднородной группы наследственных заболеваний с преимущественным поражением кожи. Они характеризуются значительным полиморфизмом клинических проявлений, подразделяются на 6 групп соответственно фенотипическим признакам и основному механизму развития. К специфическим симптомам относят очаги гипер- или гипопигментации, участки повышенного ороговения и чешуйчатости кожи, накожные опухолевые образования. План диагностики состоит из клинических, биохимических, гистологических и молекулярно-генетических методов. Лечение поддерживающее, определяется типом генодерматоза, тяжестью симптоматики.
МКБ-10

Общие сведения
Насчитывается около 200 генетически обусловленных кожных болезней, которые составляют 35% от наследственных нозологий, до 10% от всех дерматологических заболеваний. Поражение кожного покрова может быть единственным проявлением патологии, но чаще оно возникает в комплексе с мультиорганными нарушениями. Генодерматозы представляют серьезную проблему в современной дерматологии, что вызвано сложностью их лечения, отсутствием этиопатогенетической терапии, повышенным риском появления опухолей при ряде кожных заболеваний.
Причины генодерматозов
Истинные генодерматозы имеют моногенную природу — развиваются при мутации определенного гена в соматических или половых хромосомах. По характеру наследования на первом месте находится аутосомно-доминантный вариант, когда ребенку для развития болезни достаточно получить генную аномалию от одного родителя, а риск возникновения синдрома составляет 50% для младенцев обоего пола. Реже встречаются аутосомно-рецессивный, Х-сцепленный типы наследования.
При мультифакториальных генодерматозах для манифестации процесса необходимы экзогенные провоцирующие воздействия. К распространенным триггерам относят эндокринные нарушения (патологии гипоталамуса, эпифиза, щитовидной железы), хронические очаги инфекции (тонзиллит, кариес, риносинуситы), аллергические расстройства. Важными провоцирующими факторами являются повышенные психоэмоциональные нагрузки, неблагоприятная экологическая ситуация.
Патогенез
Учитывая большую вариабельность заболеваний, входящих в группу генодерматозов, выделить типичные патогенетические особенности не представляется возможным. В основе большинства пигментных нарушений лежит повышенная чувствительность клеток кожи к УФ-лучам, на фоне чего формируются очаги гиперпигментации. Генодерматозы, проявляющиеся гипопигментацией, возникают при снижении или отсутствии выработки меланина вследствие мутаций соответствующих ферментов.
Ихтиозы характеризуются нарушениями процессов кожного ороговения, замедлением отшелушивания отмерших кератиноцитов, что сопровождается утолщением и изменением внешнего вида кожных покровов. При опухолевых кожных синдромах патогенез связан с мутациями генов, отвечающих за подавление роста новообразований, в результате чего больные сталкиваются с множественными доброкачественными неоплазиями.
Классификация
В отечественной медицинской литературе распространено разделение заболеваний на 10 групп по локализации и характеру поражения. В международной дерматологической практике чаще используется классификация с учетом механизма развития и фенотипических проявлений генодерматозов, согласно которой выделяют следующие формы:
- Генодерматозы с нарушением репарации ДНК: пигментная ксеродерма, синдром Вернера, синдром Ротмунда-Томсона.
- Генодерматозы с нарушенной кератинизацией: ихтиоз, врожденный дискератоз, синдром эпителиального невуса.
- Генодерматозы с лентигинозной гиперпигментацией: синдром Пейтца-Егерса-Турена (гамартомный полипоз ЖКТ).
- Генодерматозы с кожной гипопигментацией: альбинизм, синдром Германски-Пудлака.
- Буллезные генодерматозы: буллезный эпидермолиз.
- Наследственные опухолевые синдромы: нейрофиброматоз, туберозный склероз, синдромы Кауена, Гарднера, Мюира-Торре.
В отдельную категорию выделяются мультифакториальные заболевания, которые характеризуются наследственной предрасположенностью, но для их манифестации требуется негативное влияние внешних факторов. К этой группе относят псориаз, атопический дерматит, витилиго. Большую гетерогенную группу представляют врожденные нарушения обмена веществ, которые сопровождаются кожным поражением, — болезнь Нимана-Пика, фенилкетонурия, гомоцистинурия.
Симптомы генодерматозов
Большинство заболеваний проявляются в первые месяцы после рождения ребенка. Настораживающими признаками являются патологии вынашивания беременности, преждевременные роды, симптомы задержки внутриутробного развития плода. Отдельные нозологии, например, болезнь Вернера, манифестируют у взрослых, пик их диагностики приходится на возраст 20-30 лет.
Генодерматозы с пигментными нарушениями
Для генодерматозов, протекающих с гиперпигментацией, характерны участки фотодерматита, веснушки и крупные коричневые пятна на открытых зонах тела ребенка, сухость и гиперкератоз. При прогрессировании процесса появляются участки атрофии, депигментации, деформации ушных раковин, кончика носа. У страдающих альбинизмом, напротив, отмечается неестественная бледность кожи, красноватый оттенок зрачков, отсутствие пигмента в волосах, ресницах, бровях.
Генодерматозы с нарушениями кератинизации
Генодерматозы из группы ихтиоза имеют патогномоничные проявления: обилие плотных чешуек на кожных покровах, которые придают вид змеиной кожи. Чешуйки имеют темно-коричневый цвет, плотно связаны с эпидермисом, практически не удаляются при гигиенических процедурах и трении мочалкой. Врожденный дискератоз отличается дистрофическими изменениями ногтей, лейкоплакией слизистых оболочек.
Опухолевые генодерматозы
При факоматозах (опухолевых синдромах) характерным является сочетание кожных признаков с поражением нервной системы. На теле появляются множественные болезненные новообразования темно-розового или коричневого оттенка, достигающие диаметра несколько сантиметров. По ходу нервных стволов также прощупываются многочисленные плоские опухоли. Патогномонична кожная пигментация в виде молочно-кофейных пятен.
Осложнения
Генодерматозы, как правило, сопровождаются поражением глаз по типу блефарита, кератита, эктропиона. Реже наблюдаются изъязвление роговицы, папилломы, базальноклеточный рак века. Наиболее опасным осложнением большинства опухолевых и пигментных кожных изменений является высокий риск малигнизации — образования злокачественных неоплазий. Они являются основной причиной смерти больных с генодерматозами.
При части наследственных дерматозов, например, гамартомном полипозе ЖКТ в процесс вовлекаются многие внутренние органы. Такие пациенты страдают от мультисистемных нарушений, определяющих тяжесть состояния. Обычно нозологии сочетаются с нервно-психическими расстройствами: у детей возникает синдром дефицита внимания и гиперактивности (СДВГ), расстройства аутистического спектра, проблемы с социализацией из-за нетипичной внешности.
Диагностика
Основу постановки диагноза составляет клинический осмотр пациента у детского или взрослого дерматолога. При выявлении типичных признаков (участки гипопигментации, плотные чешуйки, буллезные высыпания) удается установить предварительный диагноз, чтобы целенаправленно проводить лабораторно-инструментальную диагностику проблемы. Затем назначается комплекс диагностических мероприятий:
- Гистология кожи. Основной метод исследования, который применяется для изучения микроструктуры кожного покрова, обнаружения избыточных пигментных клеток, гиперкератоза, истончения росткового слоя. При большинстве болезней определяется отечность, воспалительная инфильтрация дермы.
- Дополнительные инструментальные методы. Зачастую генодерматозы комбинируются с соматической патологией. Поэтому больным показаны визуализационные исследования. При неопластических синдромах обязательно проводится КТ или МРТ головного мозга. Для оценки структурно-функциональных особенностей ЖКТ рекомендуются УЗИ органов брюшной полости, рентгенография.
- Биохимические анализы. При кератозах исследуется активность STS в лейкоцитах, выполняется электрофорез сывороточных протеинов для дифференцировки разных форм ихтиозов. Ценную информацию в уточнении причины генодерматозов играют иммунофлюоресцентные, иммуногистохимические методики.
- Генетическое тестирование. Обследование генома пациента — самый точный диагностический метод. Применяются методики секвенирования экзона или генома, флюоресцентной гибридизации (FISH). Поскольку исследования дорогостоящие и трудоемкие, они назначаются по строгим показаниям при невозможности установить диагноз другими способами.
Ряд моногенных кожных патологий подлежит диагностике еще во внутриутробном периоде, если родители больны или являются носителями мутантных генов. Для пренатального обнаружения генодерматозов используются ультраструктурный анализ биоптатов кожи плода, определение активности галактозидазы в амниотической жидкости, исследование уровня стероидной сульфатазы. При необходимости производится генотипирование биоматериала после амниоцентеза, биопсии хориона.
Лечение генодерматозов
Поскольку патологии вызваны мутациями в генетическом аппарате клеток, этиопатогенетического лечения не существует. Заболевания требуют пожизненной поддерживающей терапии, чтобы контролировать интенсивность кожных проявлений, нормализовать самочувствие больных, по возможности устранить неэстетичные очаги на коже. Программа лечения включает следующие направления:
- Подбор плавильной косметики. Для ухода за поврежденной кожей используются средства с кератолитиками, смягчающими и питательными компонентами, органическими кислотами. При некоторых болезнях рекомендованы топические стероиды, местные ретиноиды.
- Защита от солнца. Ультрафиолет является основным триггером развития злокачественных опухолей кожи при генодерматозах, поэтому в теплое время года обязательно наносить кремы с SPF 30-50 на всех открытых участках тела. При альбинизме нужно носить контактные линзы или очки, блокирующие УФ-лучи.
- Малоинвазивные методики. При доброкачественных опухолевых образованиях возможно их удаление с помощью электрокоагуляции, криодеструкции, лазерного излучения. Для улучшения состояния кожных покровов может назначаться дермабразия и другие косметологические процедуры.
Прогноз и профилактика
Хотя генодерматозы невозможно полностью вылечить, многие из них удается успешно контролировать поддерживающей терапией, чтобы минимизировать клинические симптомы, улучшить качество жизни больных. Менее благоприятен прогноз для пациентов с неопластическими синдромами, которые часто заканчиваются смертью от злокачественных новообразований, а также отягощении генодерматоза тяжелыми метаболическими или соматическими системными патологиями.
Учитывая наследственный характер болезней, основу профилактики составляет медико-генетическое консультирование пар из групп риска в периоде планирования зачатия. При наличии сведений о тяжелых семейных генодерматозах целесообразно проводить пренатальный скрининг либо предимплантационную генетическую диагностику (при прохождении протокола ЭКО). Для предупреждения осложнений требуется пожизненный особый уход за кожей, диспансерное наблюдение у дерматолога.
1. Факоматозы: диагностика, клиника и особенности течения различных форм заболевания/ Л. А. Юсупова, Е. И. Юнусова, З. Ш. Гараева, Г. И. Мавлютова// Лечащий врач. — 2018. — №5.
2. Федеральные клинические рекомендации. Дерматовенерология 2015: Болезни кожи. Инфекции, передаваемые половым путем. — 2016.
3. Подходы к формированию генетического прогноза развития мультифакториальных заболеваний/ А.М. Федота// Вестник биологии и медицины. — 2014. — №4.

Буллезный эпидермолиз — редкое генетическое заболевание кожи, о котором зачастую не знают не только обычные люди, но и врачи. В начале Недели буллезного эпидермолиза предлагаем полный ликбез по этому заболеванию
Буллезный эпидермолиз — это пожизненная проблема, от которой нельзя избавиться по желанию, с помощью дорогостоящего лечения или иностранных врачей. Поэтому буллезный эпидермолиз оказывает на больного сильное психологическое воздействие: наличие видимых изменений кожи и частое посещение больниц могут повлиять на социальную, учебную и профессиональную жизнь человека, значительно снизив ее качество. Сказывается и не всегда корректная реакция незнакомых людей.
Кроме того, несмотря на развитость информационных технологий, далеко не все врачи в России сегодня знают о буллезном эпидермолизе и могут поставить правильный диагноз. В итоге многих «бабочек» (так называют пациентов с диагнозом) годами «лечат» от мнимой пищевой аллергии или других заболеваний, не имеющих к реальности никакого отношения.
Чем больше общественность, в том числе и профессиональная, будет знать о буллезном эпидермолизе и связанных с ним проблемах, тем больше внимания, принятия и квалифицированной медицинской помощи будут получать такие больные. В России Неделю буллезного эпидермолиза проводит Благотворительный фонд «Дети-бабочки».

Маргарита Гехт,
ведущий врач-дерматолог благотворительного фонда «Дети-бабочки»
Что такое буллезный эпидермолиз
Буллезный эпидермолиз (БЭ) — не одно, а целая группа редких генетических заболеваний. У людей с таким диагнозом кожа и слизистые оболочки очень хрупкие. От малейшего прикосновения на них образуются пузыри. Лопаясь, они оставляют после себя болезненные раны.
Эти пузыри наверняка знакомы и вам. Такая влажная мозоль возникает на коже после целого дня, проведенного в тесных туфлях. Вечером вы нередко обнаруживаете ее уже вскрывшейся. Но если у обычных людей такие пузыри вздуваются из-за длительного трения тесной обуви и плотно прилегающей кожи, то у людей с БЭ — спонтанно или в результате малейших травм. Именно поэтому пациентов с этим заболеванием метафорично называют «бабочками». Как известно, даже легкое прикосновение к крылу бабочки снимает с него защитный слой, в результате чего она уже не может летать. При БЭ поврежденная кожа болит и может инфицироваться.
Чтобы понять, как возникает буллезный эпидермолиз, необходимо знать строение человеческой кожи.
Как устроена кожа
Кожа — самый большой орган нашего тела. Она защищает организм от микробов и аллергенов, помогает регулировать температуру тела и позволяет ощущать прикосновение, тепло и холод.
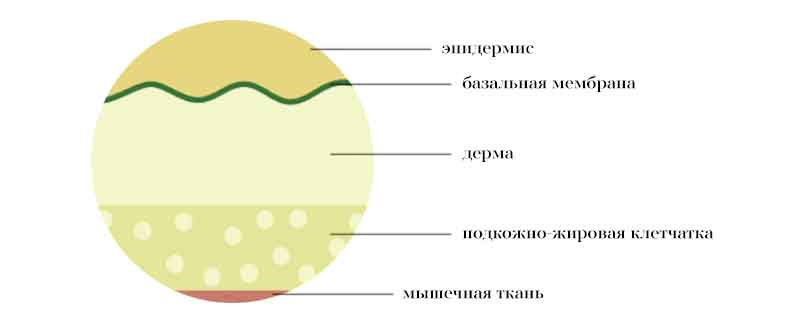
Кожа имеет три слоя:
Эпидермис
Внешний слой кожи, видимый глазу. Его основные функции — защитная и сохранения водонепроницаемого барьера — гидролипидной мантии.
Дерма
Этот слой находится под эпидермисом. Содержит плотную соединительную ткань, волосяные, сальные фолликулы, потовые железы, коллаген, эластин. В дерме проходят кровеносные сосуды и нервные окончания.
Между эпидермисом и дермой находится базальная мембрана — важная структура, соединяющая эти два слоя.
Гиподерма, или подкожно-жировая клетчатка
Глубокий слой кожи. Он состоит из жировой и соединительной ткани.
Работу, а главное, взаимодействие слоев кожи между собой определяют различные «помощники»: гены, ферменты, белки, дополнительные структуры и клетки. В случае нарушения работы хотя бы одного из «помощников» в коже происходят различные изменения. Так, при внезапно возникающих или вызванных искусственно стойких изменениях наследственных структур появляются генодерматозы, в том числе и буллезный эпидермолиз.
Что такое генодерматозы
Генодерматозы — наследственные заболевания кожи, насчитывающие несколько сотен нозологических форм и проявляющиеся различными патологическими процессами.
Причина их возникновения кроется в генах человека. Они хранят информацию, которая переводится в структуры кожи, содержащие белки, а именно — в кератин, коллаген, ламинин и интегрин, которые обеспечивают целостность и устойчивость кожи. При генетических поломках эти структуры перестают выполнять свою скрепляющую функцию, в результате кожа теряет целостность и становится хрупкой.
Буллезный эпидермолиз — один из самых тяжелых видов дерматозов данной группы.
Какие бывают виды буллезного эпидермолиза
Существует четыре основных формы буллезного эпидермолиза:
- простая;
- пограничная;
- дистрофическая;
- синдром Киндлера;
Классификация БЭ зависит от того, на каком уровне кожи произошел патологический процесс.
Простая форма
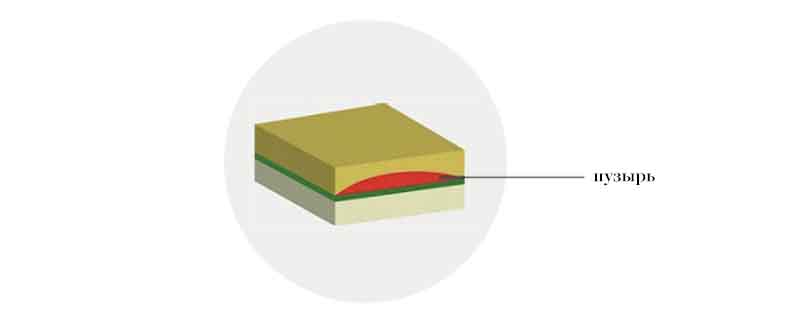
При данной форме БЭ пузыри появляются в пределах эпидермиса. Они не приводят к образованию рубцов, но причиняют сильную боль. Кроме того, летом, когда усиленное потоотделение провоцирует образование новых пузырей, простая форма БЭ всегда протекает тяжелее.
Пограничная форма
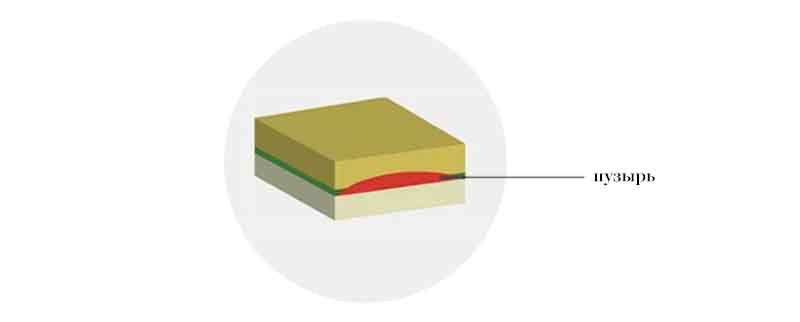
В данном случае расщепление происходит внутри базальной мембраны — структуры, которая «скрепляет» эпидермис и дерму. Более того, при определенном подтипе такой формы базальная мембрана вообще отсутствует. Из-за этого одна кожная структура может «находить» на другую, в результате чего на коже возникают многочисленные пузыри и обширные раны. Сливаясь, раны часто не оставляют на коже живого места и могут вызвать сепсис, а значит, потенциально опасны для жизни.
Дистрофическая форма
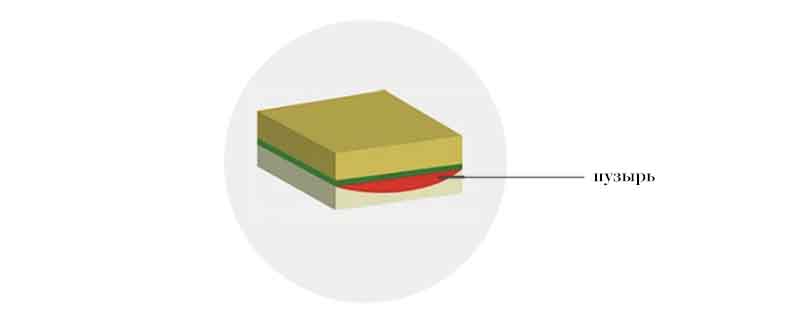
При данном типе БЭ расщепление происходит ниже базальной мембраны, в дерме. В этой области залегают ключевые структуры, которые определяют плотность и упругость кожи, — коллаген и эластин. При такой форме БЭ патологический процесс протекает именно в коллагене, в результате чего пузыри заживают с последующим образованием рубцовой ткани.
Синдром Киндлера
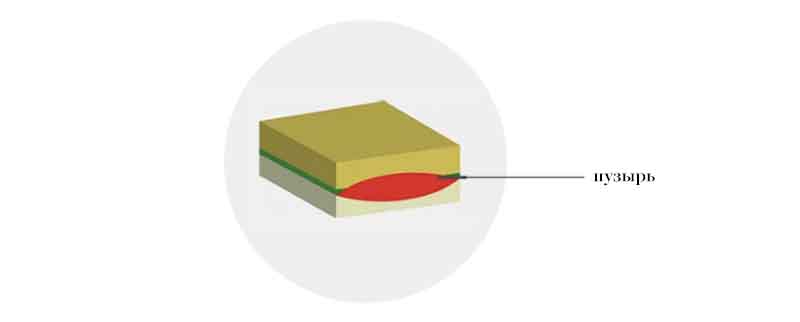
В этом случае пузыри могут образовываться одновременно на разных уровнях кожи. Кожные проявления могут быть вариабельны и зависят от степени вовлеченности гена в мутацию.
Чем отличаются врожденный и приобретенный БЭ
БЭ можно разделить на две группы — врожденный и приобретенный.
Врожденный БЭ, которому посвящена большая часть данной статьи, обусловлен генетическим дефектом. Этот дефект возникает еще внутриутробно, а сразу после рождения становятся видны внешние клинические проявления — раны на коже. Сегодня эта форма БЭ не имеет патогенетического лечения.
Приобретенный БЭ — это аутоиммунный дерматоз, связанный с выработкой специфических антител к собственному коллагену VII типа — наиболее важному компоненту фибрилл. Фибриллы — особые структуры в форме якоря, которые вместе с другими компонентами кожи скрепляют ее слои между собой.
При приобретенном БЭ образование пузырей происходит после связывания аутоантител — это антитела, которые выработались на собственные белки в результате воздействия на организм триггерных факторов (среди которых можно выделить стресс, перенесенные заболевания и оперативные вмешательства с коллагеном VII типа). Это, в свою очередь, приводит к активации каскада воспалительных реакций со стороны кожи.
Клинически заболевание характеризуется натянутыми пузырями на травмированных участках тела, которые заживают с образованием рубца. Так проявляется механобуллезная форма приобретенного БЭ. Второй наиболее частый подтип приобретенного БЭ — буллезное пемфигоидоподобное заболевание. Для него характерен кожный зуд и поражение слизистых оболочек. Для лечения приобретенного БЭ назначают местное и системное использование глюкокортикоидов и введение внутривенных иммуноглобулинов.
Как диагностируется буллезный эпидермолиз
Детальная лабораторная диагностика необходима для определения формы и подтипа БЭ и точной причины его возникновения на генетическом и белковом уровнях. Это помогает прогнозировать тяжесть состояния, оказывать надлежащую медицинскую помощь и участвовать в клинических испытаниях.
В лабораторной диагностике БЭ используются два основных метода:
1. Генетическое тестирование, которое направлено на выявление в ДНК мутации в конкретном гене, вызывающей заболевание.
2. Анализ образцов кожи с использованием методов, работающих на уровне белка, который позволяет выявить изменения в белке и компонентах кожи.
Влияет ли буллезный эпидермолиз только на кожу
Хотя физические эффекты БЭ затрагивают в основном кожу, при некоторых формах БЭ поражается не только кожа, но и слизистые оболочки внешних и внутренних органов: глаз, полости рта, пищевода, кишечника.
Заразен ли буллезный эпидермолиз
БЭ — генетическое заболевание. Это значит, что с ним рождаются. Заразиться буллезным эпидермолизом нельзя.
Как наследуется это заболевание
БЭ может быть унаследован одним из трех способов:
- аутосомно-доминантным наследованием;
- аутосомно-рецессивным наследованием;
- наследованием de novo.
Аутосомно-доминантное наследование
В этом случае один из родителей страдает заболеванием и имеет дефектный ген. Он передает измененный ген своему ребенку. Вероятность того, что любой их ребенок родится с БЭ, составляет 50%.
Аутосомно-рецессивное наследование
В этой ситуации оба родителя клинически здоровы и не имеют внешних проявлений заболевания, но являются его генетическими носителями. Чтобы их ребенок родился с БЭ, он должен унаследовать дефектный ген — часть гена от матери, часть — от отца. Возникает всем известная математическая комбинация, когда минус на минус дает плюс в виде болезни у ребенка. Вероятность того, что это произойдет, составляет 25%.
Мутации, вызывающие БЭ, которые происходят de novo
Человек, несущий вариант de novo, то есть родившийся с БЭ при здоровых родителях, в том числе не имеющих генных мутаций, имеет 50%-й шанс передать заболевание своим детям.

Можно ли вылечить БЭ
На сегодняшний день нет эффективного лечения ни одной из форм БЭ. Все доступное лечение направлено на облегчение симптомов, чтобы, в конечном счете, улучшить качество жизни человека с данным генодерматозом.
Какова продолжительность жизни пациентов с БЭ
Продолжительность жизни при буллезном эпидермолизе зависит от формы заболевания, качества ухода за кожей, профилактики внекожных проявлений, качества оказываемой медицинской помощи и прохождения ежегодных обследований. При соблюдении этих условий пациенты могут жить полноценной и качественной жизнью. В таких случаях ее продолжительность близка к продолжительности жизни людей без особенностей здоровья.
Что включает в себя облегчение симптомов БЭ
Симптоматическое лечение имеет важнейшее значение для людей с БЭ. Обычно оно включает ежедневный уход за непораженной кожей, за кожей с пузырями и ранами, а также обезболивание.
Пациентам с более тяжелыми типами БЭ требуются дополнительные процедуры, в том числе прием препаратов железа, переливание эритроцитарной массы и альбумина — белка, находящегося у данных пациентов в большом дефиците. Именно от его количества в организме зависит скорость заживления кожи, в том числе и после хирургических вмешательств. Пузыри поражают слизистую и заживают только через образование рубца, который сужает просвет органа или ограничивает его функцию. Хирургические операции позволяют освободить сросшиеся пальцы и открыть пищевод для нормального поступления пищи.
Как выглядит симптоматическое лечение при БЭ
Характер и стоимость ухода за ранами зависят от многих факторов, в том числе имеют значение:
- форма БЭ;
- возраст пациента;
- общее восприятие;
- текущее состоянии кожи;
- состояние питания;
- домашнее окружение;
- наличие качественных перевязочных материалов.
Основное лечение подразумевает использование атравматического материала, с которым при снятии не будет отходить кожа.
Основные этапы перевязки:
Тщательная подготовка к перевязке со всеми необходимыми материалами и доступным механизмом утилизации старого перевязочного материала сокращает длительность перевязки и снижает риск возникновения инфекции.
Все лица, участвующие в смене повязок, должны в обязательном порядке проводить предварительную дезинфекцию рук.
Оценка состояния кожи
Все пузыри на коже необходимо вскрывать, затем очищать образовавшиеся раны, при инфицировании — лечить.
При перевязке на кожу сначала накладывают сетчатую или губчатую повязку, затем для впитывая экссудата и излишков крема — промежуточную, после этого — фиксирующую повязку.

Что важно при выборе перевязочных средств и уходе за кожей при БЭ
Интенсивность ухода за ранами определяется в первую очередь формой БЭ. При простой форме, когда пузыри и раны возникают на отдельных участках тела, а не повсеместно, перевязка проходит гораздо быстрее, чем при более тяжелой дистрофической форме, требующей обработки повязками большей части кожи.
К счастью, в настоящее время есть множество перевязочных средств и медикаментов, которые подходят для ухода за ранами при буллезном эпидермолизе. Однако найти «правильный» материал все еще нелегко. То, что подходит для одного больного, может оказаться совсем неподходящим для другого. Поэтому даже при одной и той же форме БЭ перевязочный материал и уходовые средства подбираются индивидуально. Оценка такого соответствия должна всегда производиться совместно с врачом.
При выборе перевязочных средств необходимо также учитывать их доступность на местном уровне, ведь в разных странах для этих целей разработаны и доступны разные продукты. Кроме того, на перевязочные средства требуется значительная сумма. Так, в России на уход за кожей человека-бабочки ежемесячно нужно от 50 до 400 тыс. руб. Сегодня наше государство не всегда покрывает эти траты, а при обеспечении за счет бюджета отдает предпочтение менее дорогостоящим материалам.
Еще одним важным аспектом в лечении больного БЭ является гигиена и уход за участками кожи с рубцами, без пузырей и ран. Такой коже в первую очередь требуются восстановление и увлажнение. Чем лучше и качественнее увлажнена кожа, тем меньше выражены рубцы, меньше зуда и ниже риск инфицирования. Увлажняющие крема необходимо наносить на открытые участки кожи два-три раза в день.
Может ли легкая форма буллезного эпидермолиза перейти в тяжелую
Нет, поскольку причины, вызывающие одну форму БЭ, отличаются от причин возникновения других типов этого заболевания. Они, по сути, являются отдельными состояниями, поэтому одна форма не может перерасти в другую.
Генодерматозы — это генетически обусловленные заболевания с преимущественным поражением кожи. В настоящее время насчитывается более двухсот генетически обусловленных дерматозов, что составляет 35% от всех генетических заболеваний и примерно 10% от всех кожных болезней.
Как проявляются генетические кожные заболевания?
Некоторые генодерматозы проявляются исключительно кожной патологией, но чаще всего это комплексное поражение кожи и внутренних органов, поэтому таких пациентов должны наблюдать врачи разных специальностей.
Для современной дерматологии генетические заболевания являются серьезной проблемой, т.к. не существует терапии, воздействующей на причины появления генодерматозов, а также есть определенные сложности в лечении из-за мультисистемного поражения.
Генетические кожные заболевания не всегда проявляются сразу после рождения, многие из них манифестируют в детстве, юности и даже в более зрелом возрасте. Время появления первых симптомов болезни нередко связано с типом наследования. Одни и те же генетические заболевания могут наследоваться по-разному, что отражается и на особенностях клинической симптоматики, течении и прогнозе заболевания.
Как наследуются генетические кожные заболевания?
Генодерматозы имеют различные типы наследования, при чем доминантно наследуемые формы заболеваний нередко проявляются менее клинически выраженными расстройствами и совместимы с жизнью, в то время как рецессивно наследуемые формы этого же заболевания проявляются более тяжелой клинической симптоматикой.
Согласно классификации, предложенной Блумом (D. Bloom, 1965), генодерматозы имеют:
аутосомно-доминантный тип наследования;
аутосомно-рецессивный тип наследования;
сцепленное с полом доминантное наследование.
Какие генетические кожные заболевания встречаются чаще всего?
Самые распространенные и часто встречающиеся кожные генетические заболевания, при которых особенно четко видна роль наследственного фактора:
Буллезный эпидермолиз
Тип наследования буллезного эпидермолиза может быть как доминантным, так и рецессивным. Эпидермолиз характеризуется образованием пузырей и эрозий на коже и слизистых вследствие незначительного механического трения. Это происходит за счет нарушения межклеточных связей в эпидермисе и в дермо-эпидермальном соединении.
Клиническая картина этого генетического кожного заболевания включает в себя пузыри, эрозии и милиумы на коже, поражение ногтевых пластин (ониходистрофия), алопецию и аплазию кожи, а также ладонно-подошвенную кератодермию и пятнистую пигментацию.
Стоит отметить, что на месте крупных пузырей возникают БЭ-невусы, а эрозии и милиумы разрешаются в атрофию, рубцы и сращивание.
Лечение ВБЭ включает в себя:
заместительную терапию (переливают альбумин, кровь, плазму, иммуноглобулины);
коррекции анемий (назначают препараты железа);
антибиотикотерапию (при необходимости);
профилактику и лечение осложнений (стриктуру пищевода, остеопороз, остеопению, мониторинг новообразований);
коррекцию нутритивного статуса (исключение грубой пищи, увеличение белковых продуктов в пищевом рационе, употребление специализированных смесей для младенцев).
Помимо лечения, необходимо учитывать и основные принципы ухода за ребенком с буллезным эпидермолизом. Он заключается в следующем:
предупреждение травматизации кожного покрова — это необходимо, чтобы избежать появления новых пузырей и эрозий;
тщательный уход за эрозиями с применением современных атравматических перевязочных материалов из специальных видов ткани;
поддержание адаптивного питания для улучшения эпителизации эрозий;
психоэмоциональная поддержка как детей, так и их родителей.
Важным пунктом при уходе является подборка перевязочного материала. Основные типы классических перевязочных материалов включают в себя:
повязки на основе альгината кальция;
повязки с альгинатом и коллагеном;
повязки на основе целлюлозы;
повязки с активированным углем и импрегнацией серебра.
На данный момент продолжаются исследования на тему лечения буллезного эпидермолиза, и существуют несколько перспективных методов, которые пока что являются экспериментальными. К ним относится клеточная, генная и заместительная белковая терапии.
Ихтиозы
Ихтиозы — группа наследственных генетических кожных заболеваний, характеризующихся генерализованным нарушением ороговения кожного покрова по типу гиперкератоза.
Предположительная частота встречаемости — 0,15–0,2%, то есть 1:300 тысяч человек. В 77,5% случаев это первые дети в семье, рожденные у молодых родителей (22–25 лет). Соотношение заболеваемости по полу ж:м = 1,55:1,0.
В международной классификации болезней ихтиозы классифицируются на:
ихтиоз, связанный с Х-хромосомой;
врожденная буллезная ихтиоформная эритродермия;
ихтиоз плода, более известный как «плод Арлекин».
Каждый вид ихтиоза имеет свою специфическую клиническую картину, но существуют и общие проявления, такие как эритродермии, гиперкератозы, шелушение, контрактуры суставов, дистрофии ногтей и неприятный запах от пациента.
Основой ухода при данном генетическом кожном заболевании в первый год жизни ребенка является применение ванн с добавками масла, а после трех лет поочередно применяют ванны с маслом и с пищевой содой.
Также важной составляющей ухода является механическое удаление выраженных участков кератоза. После 10–20 минут размягчения кератозов в ванне необходимо использовать шелковые лоскуты или мочалки-рукавички для мягкого удаления элементов. В среднем, уход за всем телом при тяжелом ихтиозе занимает 60–90 минут в день.
Исходя из современных исследований, для купирования зуда и уменьшения ксероза кожи рекомендовано применение 10% парафина и 15% глицерола, а также кремы с 10% мочевиной в составе. В качестве альтернативы используют местные ретиноиды (0,05% ретиноевая мазь), а в случае присоединения вторичной инфекции применяют наружные антисептики.
Тяжелые формы ихтиоза направляют на стационарное лечение, где назначают системные глюкокортикостероиды, ретиноиды, внутривенное введение иммуноглобулинов и проводят дезинтоксикационную терапию.
Нейрофиброматоз
Нейрофиброматоз I типа (Болезнь Реклингхаузена) является самым распространенным из всех аутосомно-доминантных заболеваний, с частотой встречаемости 1:2500–1:30000. Лица мужского пола страдают данным генодерматозом чаще женщин.
Диагноз ставится на основании клинической картины и при наличии не менее двух нижеперечисленных критериев:
Пятна цвета «кофе с молоком» на коже. При количестве 6 и более таких пятен в размере до 0,5 см, в совокупности с другими критериями можно предполагать данный генодерматоз.
Две и более нейрофибромы любого типа. Нейрофибромы — это мягкие узлы на коже различного размера и цвета (от телесного до коричневого).
Веснушчатость в интертригинозных участках. Наличие мелких гиперпигментрированных пятен на таких участках, как подмышечные впадины, паховая область, под молочными железами у женщин.
Глиома зрительного нерва — опухоль глиальных клеток зрительного нерва.
Узелки Лише — коричневые пигментные пятнышки на радужной оболочке. Наличие двух и более узелков является диагностическим критерием НФ-I.
Изменение костей (дисплазия клиновидной кости, истончение длинных трубчатых костей с псевдоартрозами и др.).
Наличие нейрофиброматоза у родственников первой степени родства.
Стоит отметить, что общее количество пятен не коррелирует с тяжестью НФ, а узелки Лиша не влияют на функцию зрения.
Пациенты с данным генетическим кожным заболеванием наблюдаются у ряда специалистов — генетика, офтальмолога, дерматолога и других, но, к сожалению, лечение нейрофиброматоза все еще остается невозможным.
Иногда выполняют полную резекцию элементов хирургическим путем либо частичную резекцию с помощью лазера СО2. В некоторых случаях назначают химио- либо лучевую терапию.
Тактику ведения пациента выбирает консилиум врачей, исходя из формы, симптомов и степени тяжести нейрофиброматоза.
Многие генодерматозы выглядят устрашающе, да и само словосочетание «генетическое кожное заболевание» пугает родителей. Но важно понимать, что при вовремя поставленном диагнозе, своевременной терапии и правильном уходе, прогнозы многих генодерматозов благоприятные.
Список литературы
Беленький Г. Б. Генетические факторы в дерматологии, М., 1970; Суворова К. Н. Генодерматозы, ч. 1–2, М., 1969–1972;
Butterworth Т. a. Streаn L. P. Clinical geno dermatology, Baltimore, 1962; Desmons F. et Walbaum R. Les affections cutan£es h6r£ditaires liees au sexe, Lille med., t. 17, p. 447, 2002;
Кубанов А. А., Альбанова В. И., Карамова А. Э. и др. Распространенность врожденного буллезного эпидермолиза у населения Российской Федерации // Вестн. дерматологии и венерологии. 2015. № 3. С. 21-30.
Снарская Е. С., Кряжева С. С., Карташова М. Г., Бобров М. А., Филатова И. В. Врожденный дистрофический гиперпластический буллезный эпидермолиз Коккейна — Турена // Рос. журн. кожных и венерич. болезней. 2011. № 5. С. 34-41.
Коталевская Ю. Ю., Марычева Н. М. Буллезный эпидермолиз: основные клинические проявления // Педиатрия. № 4. 2014. С. 70-72.
Kelsell D.P., Norgett E.E., Unsworth H. et al. Mutations in ABCA12 underlie the severe congenital skin disease harlequin ichthyosis // Am. J. Hum. Genet., 2005. Vol. 76. № 5 Р. 794-803.
Клинические рекомендации Союза педиатров России. Ихтиоз у детей. М., 2016. С. 39
Тальникова, Е.Е. Ихтиоз: к вопросу наследования (обзор) / Е.Е. Тальникова, В.Н. Шерстнева, А.В. Моррисон, С.Р. Утц // Саратовский научно-медицинский журнал. — 2016. — № 12(3). — С. 513-517
Читайте также: