
Биопсия кожи при склеродермия
Обновлено: 25.04.2024
а) Клиника. Диагноз системного склероза и сопутствующих заболеваний основывается, прежде всего, на характерных клинических признаках. Поражение кожи характеризуется различной степенью ее утолщения и уплотнения. Могут отмечаться изменения кожной пигментации, в частности крапчатая гиперпигментация по типу «соли с перцем».
Другие важные кожные проявления:
• В начальных стадиях заболевания - кожный зуд и отечность.
• Склеродактилия.
• Язвочки и вдавления на кончиках пальцев.
• Телеангиэктазии.
• Кальциноз кожи.
Линейная форма ограниченной склеродермии на лбу, называемая «En coup de sabre» - «удар саблей» Линейная форма ограниченной склеродермии («удар саблей»), появившаяся три месяца назад на лбу у 41-летней женщины латиноамериканкого происхождения Склеродермия со склеродактилией - плотной блестящей кожей на пальцах Склеродактилия с сужением пальцев и крапчатой гиперпигментацией Тяжелая системная склеродермия с деформацией кистей как результат склеродактилии, приводящей к тяжелым сгибательным контрактурам
Диагноз ограниченной склеродермии основывается на наличии типичного утолщения и уплотнения кожи, ограниченного одним очагом. Диагноз системного склероза предполагается при типичном утолщении и уплотнении (склероз) кожи, не ограниченном одним участком (то есть не является локализованной склеродермией). Комбинация кожных симптомов с одним или более типичным системным признаком свидетельствует в пользу диагноза системного склероза.
Согласно Критериям Американской ревматологической ассоциации для диагноза системного склероза требуется наличие одного главного (большого) или двух дополнительных (малых) критериев.
• Главными критериями являются типичные склеродермические изменения кожи: напряженность, утолщение, плотная отечность, не оставляющая следов после вдавливания, а также (за исключением локализованных форм склеродермии):
- Склеродактилия: вышеописанные изменения ограничены пальцами кистей и стоп.
- Проксимальная склеродермия: описанные выше изменения определяются проксимальнее запястно-фаланговых или запястно-пястных суставов, а также на других участках конечностей, лице, шее или туловище (грудь или живот) и практически всегда включают склеродактилию.
• Дополнительные (малые) критерии склеродермии:
- Вдавленные рубчики или потеря ткани на подушечках пальцев кистей.
- Двусторонний отек пальцев кистей или ладоней.
- Аномальная пигментация кожи: часто гиперпигментация с очагами точечной или пятнистой гипо-пигментации.
- Феномен Рейно.
- Двусторонний базилярный фиброз легких.
- Неподвижность нижнего отдела пищевода.
- Образование выпячиваний в ободочной кишке: дивертикулы ободочной кишки с широкими устьями расположены вдоль противобрыжеечного края.
б) Анализы при склеродермии. Характерным для системной склеродермии является положительный тест AHA с пятнистым, гомогенным или нуклеолярпым окрашиванием. С ОКСС часто ассоциируются анти-центромерные антитела. Анти-ДНК топоизомеразы (Scl-70) антитела являются высоко специфичными для системного склероза и связанных с ним интерстициальных заболеваний легких и почек. Несмотря на невысокую чувствительность, антитела к анти-РНК полимеразе I и III являются специфическими для системного склероза. Обычно при дисфункции специфического органа проводятся и другие виды тестирования.
Для диагностики ограниченной и системной склеродермии может использоваться биопсия.
Системная склеродермия с поражением голеней и мышечной атрофией у пациента, представленного на рисунке выше Системная склеродермия с телеангиэктазиями и некрозом кончиков пальцев Телеангиэктазии на лице у пациента, представленного на рисунке выше Феномен Рейно с тяжелой ишемией, приводящей к некрозу кончиков пальцев Кальциноз над локтевым суставом у пациента с CREST-синдромом Системная склеродермия с крапчатой гиперпигментацией. Внешний вид кожи напоминает картину «соли с перцем»
в) Дифференциальная диагностика склеродермии:
• Идиопатические случаи заболеваний, которые ассоциируются с системным склерозом, такие как феномен Рейно, почечная недостаточность и гастроэзофагеальный рефлюкс.
• Системная красная волчанка представлена системными симптомами и типичной сыпью, которая может рубцеваться. Тестирование на антинуклеарные антитела (AНA) обычно помогает установить диагноз.
• Дискоидная красная волчанка представлена локализованными бляшками, которые рубцуются. Для установления диагноза обычно выполняется биопсия.
• Микседема ассоциируется с гипотиреоидизмом и характеризуется утолщением и огрублением кожи. Исследования уровней гормонов щитовидной железы, как привило, подтверждают диагноз.
• Амилоидоз кожи может проявляться утолщением и ригидностью кожи. В биоптате кожи определяется амилоидный инфильтрат.
• Грибовидный микоз представлен пятнами и бляшками с фиолетовым оттенком по всему кожному покрову. Диагноз обычно подтверждается при биопсии.
г) Лечение склеродермии:
• Очаги локализованной склеродермии, в том числе ограниченной (морфеа) склеродермии размягчаются после терапии УФ-лучами диапазона А. Другие средства лечения включают местные кортикостероиды высокой степени активности и местный кальципотриол. Прием метотрексата начинают с 7,5 мг внутрь в неделю, в дальнейшем дозировка подбирается по необходимости. С успехом применяется комбинация высоких доз системных кортикостероидов и низких доз метотрексата.
• Для симптоматической терапии может применяться противозудное лечение смягчающими местными препаратами, блокаторами гистамина 1 (Н1) и гистамина 2 (Н2), пероральным доксепином и малыми дозами пероральных глюкокортикостероидов.
• Телеангиэктазии можно замаскировать декоративной косметикой или лечить лазером.
• При симптомах феномена Рейно могут помочь блокаторы кальциевых каналов, прасозин, производные простагландина, дипиридамол, аспирин и местные нитраты. У пациентов с первичной болезнью Рейно показал свою эффективность силдепафил (20 мг перорально два раза в день). Пациентам рекомендуется избегать холода, стресса, приема никотина, кофеина, препаратов с симпатомиметическим противоотечным действием. При гастроэзофагеальном рефлюксе могут эмпирически применяться препараты, снижающие кислотность. При затрудненном глотании , могут быть полезны прокинетические препараты.
• Некоторые локализованные очаги могут быть иссечены.
• Неутверждеппые методы терапии при поражениях кожи включают интерферон-гамма, микофенолата мофетил (1—1,5 г перорально в день) и циклофосфамид (50-150 мг/день внутрь однократно). Распространенное кожное заболевание экспериментально лечили Д-пеницилламином (250-1500 мг/день перорально 2-3 раза в день перед едой натощак).
• Главное направление лечения при поражении почек - контроль артериального давления с применением в качестве препаратов первой линии ингибиторов ангиотензин-копвертирующего фермента (АКФ). По показаниям применяется гемодиализ или перитонеальный диализ.
• Средства лечения легочной гипертензии, ассоциированной с системной склеродермией, включают антагонист эндотелиальпого рецептора-бозентан (62,5 мг перорально два раза в день в течение 4 недель, затем повышают до 125 мг перорально два раза в день), силденафил - ингибитор фосфодиэстеразы-5 и различные аналоги простациклина (эпопростеиол, трепростинил и илопрост). При легочном фиброзирующем альвеолите может применяться циклофосфамид.
• Миозит лечат стероидами, метотрексатом и азатиоприном (50-150 мг/день). Дозы преднизона свыше 40 мг/день повышают частототу склеродермальных почечных кризов. Артралгии можно лечить ацетаминофеном и нестероидными противовоспалительными препаратами.
д) Рекомендации пациентам со склеродермией. Пациенту необходимо избегать травм кожи (особенно пальцев) и воздействия холода, а также воздержаться от курения. Пациент должен знать о потенциальных осложнениях и самостоятельно следить за признаками и симптомами прогрессировать заболевания.
е) Наблюдение пациента врачом. Пациентам с системным склерозом требуется регулярное наблюдение врача, не реже чем каждые 3-6 месяцев для оценки активности и прогрессировать заболевания.
ж) Список использованной литературы:
1. Lawrence RC, Helmick CG,Arnett PC, et al. Estimates of the prevalence of arthritis and selected musculoskeletal disorders in the United States. Arthritis Rheum. 1998;41:778.
2. Akesson A,Wollheim FA. Organ manifestations in 100 patients with progressive systemic sclerosis:A comparison between the CREST syndrome and diffuse scleroderma. Br J Rheumatol. 1989;28:281.
3. Medsger ТА, Jr., Masi AT. Survival with scleroderma. II. A lifetable analysis of clinical and demographic factors in 358 male U.S. veteran patients. J Chronic Dis. 1973;26:647.
4. Tuffanelli DL,Winklemann RK. Systemic scleroderma. A clinical study of 727 cases. Arch Dermatol. 1961;84:359.
5. Janosik DL, Osborn TG, Moore TL, et al. Heart disease in systemic sclerosis. Semin Arthritis Rheum. 1989;19:191.
6. Byers RJ, Marshall DA, Freemont AJ. Pericardial involvement in systemic sclerosis. Ann Rheum Dis. 1997;56:393.
7. American Rheumatism Association Diagnostic and Therapeutic Criteria Committee. Preliminary criteria for the classification of systemic sclerosis (scleroderma). Subcommittee for scleroderma criteria of the American Rheumatism Association Diagnostic and Therapeutic Criteria Committee. Arthritis Rheum. 1980;23(5): 581-590.
8. Reveille ID, Solomon DH. Evidence-based guidelines for the use of immunologic tests: Anticentromere, Scl-70, and nucleolar antibodies. Arthritis Rheum. 2003;49:399.
9. Kreuter A, Breuckmann F, Uhle A, et al. Low-dose UVA1 phototherapy in systemic sclerosis: Effects on acrosclerosis. / Am Acad Dermatol. 2004;50:740.
10. Seyger MM, van den Hoogen FH, de Boo T, de Jong EM. Lowdose methotrexate in the treatment of widespread morphea. J Am Acad Dermatol. 1998;39:220.
11. Kreuter A, Gambichler T, Breuckmann F, et al. Pulsed high-dose corticosteroids combined with low-dose methotrexate in severe localized scleroderma. Arch Dermatol. 2005; 141:847.
12. Thompson AE, Shea B,Welch V, Fenlon D, Pope JE. Calciumchannel blockers for Raynaud’s phenomenon in systemic sclerosis. Arthritis Rheum. 2001;44:1841.
13. Clifford PC, Martin MF, Sheddon EJ, Kirby JD, Baird RN, Dieppe PA.Treatment of vasospastic disease with prostaglandin El. Br Med J. 1980;281:1031.
14. Fries R, Shariat K, von Wilmowsky H, Bohm M. Sildenafil in the treatment of Raynaud’s phenomenon resistant to vasodilatory therapy. Circulation. 2005; 112:2980.
15. Falanga V, Medsger ТА, Jr. D-penicillamine in the treatment of localized scleroderma. Arch Dermatol. 1990; 126(5):609-612.
16. Tashkin DP, Elashoff R, Clements PJ, et al. Cyclophosphamide versus placebo in scleroderma lung disease. N Engl J Med. 2006;354:2655.
17. Steen VD, Medsger ТА, Jr. Case-control study of corticosteroids and other drugs that either precipitate or protect from the development of scleroderma renal crisis. Arthritis Rheum. 1998;41:1613.
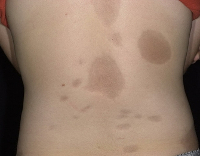
Очаговая склеродермия – это хроническое заболевание соединительной ткани, характеризующееся преимущественным поражением кожных покровов. Клинически проявляется уплотнением (индурацией) различных участков кожи с последующей атрофией и изменением пигментации, образованием контрактур. Диагноз ставится на основании симптоматики, обнаружения в крови антинуклеарного фактора и антицентромерных антител. В сомнительных случаях проводится гистологическое исследование кожи. Лечение заключается в применении глюкокортикостероидов, иммунодепрессантов, антифиброзных средств, блокаторов кальциевых каналов и проведении ПУВА-терапии. В ряде случаев выполняются хирургические операции.
МКБ-10
Общие сведения
Очаговая (локализованная, ограниченная) склеродермия – хроническое аутоиммунное заболевание из группы диффузных болезней соединительной ткани. Патология встречается повсеместно, распространенность составляет от 0,3 до 3 случаев на 100 000 человек. Чаще страдают женщины европеоидной расы. Возраст манифестации очаговой склеродермии зависит от формы. Бляшечная склеродермия чаще встречается у взрослых (30-40 лет), линейная - у детей от 2 до 14 лет, склероатрофический лихен – у женщин старше 50 лет. При локализованной форме, в отличие от системной, поражение внутренних органов в большинстве случаев либо минимально, либо отсутствует. Имеется ассоциация склеродермии с патологиями щитовидной железы (тиреоидитом Хашимото, болезнью де Кервена).
Причины
Точная причина заболевания неизвестна. Предполагается этиологическая роль бактерии Borrelia burgdorferi, вызывающей лайм-боррелиоз, однако убедительных данных за эту теорию на сегодняшний день нет. В развитии склеродермии важную роль играет наследственная предрасположенность. Были выявлены более частые случаи очаговой склеродермии среди близких родственников. При проведении генетических исследований обнаружена взаимосвязь между определенными генами гистосовместимости (HLA – DR1, DR4) и локализованной формой заболевания. Провоцирующими факторами, способствующими возникновению склеродермии, являются переохлаждения, травмы, постоянные вибрационные воздействия на кожу, прием лекарственных препаратов (блеомицина). Триггерными эффектами также обладают различные химические соединения (хлорвинил, кремний, нефтепродукты, сицилий, эпоксидная смола, пестициды, органические растворители).
Патогенез
Выделяют три основных патогенетических механизма склеродермии – фиброз (разрастание соединительной ткани), аутоиммунное повреждение и сосудистые нарушения. Иммунная аутоагрессия заключается в выработке лимфоцитами антител к соединительной ткани и ее компонентам. Также лимфоциты синтезируют интерлейкины, которые стимулируют пролиферацию фибробластов, гладкомышечных клеток и образование коллагена. Разрастающаяся при этом соединительная ткань замещает нормально функционирующую ткань. В результате повреждения эндотелия сосудов антителами и пролиферирующими гладкомышечными клетками снижается уровень простациклина (вещества, обладающего антиагрегантными и вазодилатирующими свойствами). Это приводит к спазму микрососудов, повышению адгезии и агрегации форменных элементов крови, внутрисосудистой коагуляции и микротромбозу.
Классификация
Очаговая склеродермия подразделяется на множество форм. Наиболее распространенными являются бляшечная и линейная. У ряда пациентов могут наблюдаться одновременно несколько вариантов заболевания. Существует целый ряд классификаций, но наиболее оптимальной и широко используемой считается классификация клиники Мэйо, включающей следующие разновидности очаговой склеродермии:
- Бляшечная. Данная форма в свою очередь подразделяется на поверхностную (морфеа) и узловатую (келоидоподобную). Характерны типичные участки уплотнения кожи с атрофией и нарушением пигментации.
- Линейная. К ней относятся полосовидная, саблевидная формы, а также прогрессирующая гемиатрофия лица Парри-Ромберга. Очаги располагаются в виде линий по ходу сосудисто-нервного пучка.
- Генерализованная (многоочаговая). Проявляется сочетанием бляшечного и линейного вариантов. Очаги распространены по всему телу.
- Буллезная. При данной разновидности на коже возникают пузыри с жидкостным содержимым, оставляющие после себя эрозии.
- Пансклеротическая инвалидизирующая. Наиболее неблагоприятная форма очаговой склеродермии. Характеризуется тяжелым, прогрессирующим течением, плохо поддается лечению. Поражаются все слои кожи и ткани, лежащие под ней. Развиваются грубые контрактуры суставов и длительно незаживающие язвы на коже.
- Склероатрофический лихен Цумбуша (болезнь белых пятен). Характерно образование пятен белого цвета, сопровождающихся нестерпимым зудом. Преимущественная локализация пятен – половые органы.
Симптомы
Для клинической картины типично образование на коже очагов, которые проходят три последовательных стадий развития – отек, индурацию (уплотнение) и атрофию. В начале заболевания на коже конечностей, шеи или туловища появляются пятна сиреневого или лилового цвета, имеющие нечеткие края. Размер пятен может сильно варьировать – от просяного зерна до размеров ладони и больше. На этом этапе пациент не испытывает каких-либо неприятных ощущений или боли. Затем пятна начинают отекать, кожа в центре очага уплотняется, становится блестящей, приобретает цвет слоновой кости. Пациент начинает ощущать зуд, покалывания, стянутость кожи, болезненность. Далее наступает стадия атрофии. Кожа в очагах истончается, прекращается рост волос, нарушается потоотделение, возникает стойкая дисхромия (гипер- или депигментация) и телеангиэктазии. Иногда развивается атрофодермия (участки западения кожи).
При линейной склеродермии очаги расположены по ходу нервов и сосудов. В случае локализации на коже лица очаги по внешнему виду напоминают рубец от удара саблей (саблевидная форма). Прогрессирующая гемиатрофия представляет собой глубокий процесс с поражением всех тканей половины лица - кожи, подкожной клетчатки, мышц и костей лицевого скелета, что приводит к выраженной деформации лица, обезображивающей внешний вид пациента. Также происходит атрофия половины языка и снижение вкусовой чувствительности.
Из внекожных признаков очаговой склеродермии стоит отметить офтальмологические и неврологические проявления при гемиатрофии Парри-Ромберга. Они включают выпадение ресниц и бровей на стороне поражения, западение глазного яблока из-за атрофии глазных мышц и орбитальной клетчатки, нейропаралитический кератит, головокружения, когнитивные нарушения, мигренозные головные боли, эпилептические припадки. Также возможно развитие феномена Рейно. Симптомы синдрома Рейно следующие – стадийное изменение окраски кожи пальцев рук вследствие вазоспазма и последующей гиперемии (бледность, цианоз, покраснение), сопровождающееся онемением, болью и покалыванием в пальцах рук. Остальные экстрадермальные проявления, характерные для системной склеродермии, встречаются крайне редко.
Осложнения
Наиболее распространенная проблема рассматриваемого заболевания – косметические дефекты. Серьезные осложнения, представляющие угрозу для жизни больного, возникают редко. К ним относятся нарушение мозгового кровообращения при гемиатрофии лица, ишемия и гангрена пальцев рук при феномене Рейно, выраженные контрактуры суставов, инвалидизирующие пациента. Через несколько лет после дебюта болезни могут развиться тяжелые поражения внутренних органов – фиброз легких, легочная гипертензия, фиброз миокарда, перикардит, стриктуры пищевода, острая нефропатия, почечная недостаточность.
Диагностика
Пациентов с очаговой склеродермией курируют врачи ревматологи и дерматологи. При постановке диагноза учитывается клиническая картина, семейный анамнез. Все методы диагностики направлены в первую очередь на определение степени вовлечения внутренних органов и исключение системной склеродермии. С этой целью применяются следующие исследования:
- Лабораторные. В анализах крови выявляются эозинофилия, повышение уровня ревматоидного фактора, гаммаглобулинов, высокие титры антицентромерных антител и антинуклеарного фактора (АНФ). Наличие антител к топоизомеразе (анти-Scl 70) свидетельствует в пользу системного процесса. При развитии «склеродермической почки» в моче появляются белок и эритроциты.
- Инструментальные. При капилляроскопии наблюдается дилатация капилляров без участков некроза. По данным ФЭГДС могут встречаться признаки эзофагита, стриктуры пищевода. При фиброзе миокарда на ЭКГ иногда обнаруживаются нарушения ритма сердца, на ЭхоКГ – зоны гипокинеза, выпот в перикардиальную полость. На рентгенографии или компьютерной томографии легких отмечаются интерстициальные изменения.
- Гистологическое исследование биоптата кожи. Заключительный этап, позволяющий достоверно поставить диагноз. Проводится при сомнительных результатах предыдущих исследований. Характерны следующие признаки - инфильтрация лимфоцитами, плазмоцитами и эозинофилами в ретикулярном слое дермы, утолщенные коллагеновые пучки, набухание и склероз сосудистой стенки, атрофия эпидермиса, сальных и потовых желез.
Очаговую склеродермию дифференцируют с другими формами склеродермии (системной, склеродермой Бушке), дерматологическим заболеваниями (саркоидозом кожи, липонекробиозом, склеродермоподобной формы поздней кожной порфирии, базально-клеточным раком), поражением мягких тканей (панникулитом, липодерматосклерозом, эозинофильным фасциитом). В дифференциальной диагностике принимают участие онкологи, гематологи.
Лечение
Этиотропной терапии не существует. Метод лечения и вид лекарственного средства необходимо подбирать с учетом формы заболевания, тяжести течения и локализации очагов. При линейной и бляшечной формах используются топические глюкокортикостероиды высокой и сверхвысокой активности (бетаметазон, триамцинолон), синтетические аналоги витамина Д. При выраженной индурации кожи эффективны аппликации с диметилсульфоксидом. В случае поражений внутренних органов с целью уменьшения фиброзообразования назначаются пеницилламин и инъекции гиалуронидазы.
При неглубоких процессах хорошим терапевтическим действием обладает ПУВА-терапия, которая включает облучение кожи ультрафиолетовыми волнами длинного спектра с одновременным пероральным или наружным применением фотосенсибилизаторов. Тяжелое поражение кожи служит показанием к применению иммунодепрессантов (метотрексата, такролимуса, микофенолата), синдром Рейно - блокаторов кальциевых каналов (нифедипина) и препаратов, улучшающих микроциркуляцию (пентоксифиллина, ксантинола никотината). При склероатрофическом лихене проводится низкоинтенсивная лазеротерапия. В случае развития контрактур суставов, значительно затрудняющих движения, или грубых деформаций скелета и косметических дефектов лица требуется хирургическая операция.
Профилактика и прогноз
В подавляющем большинстве случаев очаговая склеродермия имеет доброкачественное течение. Правильно подобранная терапия позволяет добиться регресса симптомов. Иногда наступают спонтанные ремиссии заболевания. Неблагоприятные исходы возникают при тяжелых формах (прогрессирующей гемиатрофии лица, пансклеротической инвалидизирующей склеродермии), а также поражении внутренних органов. Эффективных методов профилактики не разработано. Рекомендуется избегать или максимально ограничить контакт кожи с химическими соединениями (кремнием, сицилием, хлорвинилом, нефтепродуктами, органическими растворителями, пестицидами, эпоксидной смолой).
2. Ревматические заболевания/ Под ред. Дж.Х. Клиппела, Дж.Х. Стоуна, Л.Дж. Кроффорд, П.Х. Уайт – 2012.
3. Диффузные болезни соединительной ткани: руководство для врачей/ под ред. проф. Мазурова В.И. –2009.
Авторы: Ахмина Н.И. 1 , Уткин П.С. 2 , Шалатонин М.П. 3 , Чабаидзе Ж.Л. 1 , Дементьев А.А. 1 , Заплатников А.Л. 1
1 ФГБОУ ДПО РМАНПО Минздрава России, Москва, Россия
2 ГБУЗ ДИКБ No 6 ДЗМ, Москва, Россия
3 ГБУЗ «Морозовская ДГКБ ДЗМ», Москва, Россия
Склеродермия — редкая патология в педиатрической практике, хотя и занимает 3-е место после ювенильного идиопатического артрита и системной красной волчанки среди всех ревматических заболеваний детского возраста. При этом варианты врожденной склеродермии (ВС) рассматриваются как казуистика, так как в мировой литературе до настоящего времени имеются лишь единичные описания таких случаев. Этиология ВС неизвестна. Основными патогенетическими звеньями заболевания являются: повреждение сосудистого русла, дисфункция иммунной системы с нарушением регуляции высвобождения цитокинов и измененный синтез коллагена с пролиферацией фибробластов и последующим развитием фиброза. В статье приведено описание клинического случая склеродермии у новорожденного ребенка с врожденной локализованной нодулярной формой, клинические проявления которой были выявлены уже при рождении. Описаны трудности верификации диагноза на основании клинических и рутинных лабораторно-инструментальных исследований. Подчеркивается роль современных морфологических исследований, ввиду отсутствия специфических лабораторных тестов, в установлении диагноза склеродермии у новорожденного ребенка. Также представлен краткий обзор литературы, посвященной проблеме ВС.
Ключевые слова: врожденная склеродермия, дети, диагностика, дифференциальная диагностика, морфологические исследования.
N.I. Akhmina 1 , P.S. Utkin 2 , M.P. Shalatonin 3 , Zh.L. Chabaidze 1 , A.A. Dement’ev 1 , A.L. Zaplatnikov 1
1 Russian Medical Academy of Continuous Professional Education, Moscow,
2 Children’s Infectious Clinical Hospital No. 6, Moscow, Russian Federation
3 Morozov Children’s City Clinical Hospital, Moscow, Russian Federation
Scleroderma is a rare condition in pediatrics although it ranks third (after juvenile idiopathic arthritis and systemic lupus erythematosus) among rheumatic diseases in children. Congenital scleroderma is considered casuistic since only single case reports are available in publications worldwide. The etiology of congenital scleroderma remains elusive. The principal pathogenic mechanisms are vascular network impairment, immune system dysfunction with abnormal regulation of cytokine release and altered collagen synthesis with fibroblast proliferation and fibrosis. This paper describes a newborn girl with congenital localized nodular scleroderma whose clinical presentations were detected at birth. The authors discuss difficulties with the diagnosis based on clinical and routine lab and instrumental tests. The role of modern morphological studies (given the lack of specific lab tests) to diagnose scleroderma in a newborn is emphasized. Finally, this paper provides a brief review of published data on congenital scleroderma.
Keywords: congenital scleroderma, children, diagnostics, differential diagnosis, morphological studies.
Введение
Ювенильной склеродермией (ЮС) называют склеродермию, при которой клинические проявления манифестируют в возрасте до 18 лет. ЮС является редкой ревматологической патологией детского возраста. В то же время среди всех аутоиммунных заболеваний соединительной ткани у детей ЮС занимает 3-е место после ювенильного идиопатического артрита и системной красной волчанки. Этиология ЮC остается неизвестной. Основными патогенетическими звеньями заболевания являются: повреждение сосудистого русла, дисфункция иммунной системы с нарушением регуляции высвобождения цитокинов и измененный синтез коллагена с пролиферацией фибробластов и последующим развитием фиброза. Специфические лабораторные тесты, на основании которых можно было бы верифицировать заболевание, до настоящего времени не разработаны. Диагноз устанавливается, как правило, на основании клинико-анамнестических данных и с учетом характерных гистологических признаков, если были проведены морфологические исследования. Учитывая клинические особенности, выделяют системную и ограниченную ЮС. Системная ЮС характеризуется развитием склероза кожи и поражением внутренних органов. При ограниченной ЮС имеет место преимущественное поражение кожи и/или подлежащих тканей с развитием в дальнейшем контрактур и значительно реже — с повреждением костных структур [1–5]. Особо следует отметить, что в мировой литературе [6–11] имеются описания единичных случаев склеродермии, клинические проявления которой были обнаружены уже при рождении детей (врожденная склеродермия, ВС). Казуистическая частота встречаемости ВС и трудности ее диагностики явились основанием для настоящей публикации.
Клиническое наблюдение
Девочка М. родилась от 39-летней женщины, от 5-й беременности, от 3-х оперативных родов на 39-й неделе гестации. Предыдущие беременности: 1-я — срочные роды; 2-я и 3-я — медицинские аборты; 4-я — оперативные своевременные роды; 5-я — настоящая беременность беременность, которая протекала с токсикозом в I триместре, острой респираторной инфекцией без подъема температуры во II триместре и преэклампсией 1-й степени в III триместре.
В семейном анамнезе: у матери имеются желчнокаменная болезнь, миопия слабой степени, варикозная болезнь вен нижних конечностей, эрозия шейки матки. Во время беременности мать в плановом порядке обследована на сифилис, гепатиты В и С, ВИЧ-инфекцию: результаты отрицательные. Отцу 40 лет, курит. Заболевания кожи и онкологическая патология в семейном анамнезе не установлены. Аллергический анамнез не отягощен.
При рождении: масса 3100 г, длина 50 см, окружность головы 35 см, окружность груди 32 см. Оценка по шкале Апгар 7–8 баллов. Состояние ребенка при рождении средней степени тяжести. При рождении на туловище (грудь, живот, спина), на конечностях и на лице отмечены узловатые образования от 3–5 мм до 25–35 мм в диаметре, плотные, возвышающиеся над уровнем кожи, местами сливающиеся, кожа над ними гиперемирована с синюшным оттенком (рис. 1). Дыхательные и гемодинамические нарушения отсутствовали. В неврологическом статусе — синдром угнетения с элементами возбуждения.

На 2-е сутки жизни девочка переведена из роддома в ГБУЗ ДИКБ № 6 ДЗМ. При поступлении, помимо описанных элементов на коже, был отмечен также гепатолиенальный синдром: печень (по Курлову) +1,5 см, +3,0 см, +1 см; селезенка +2 см. Рефлексы новорожденного вызывались в полном объеме. Учитывая клинические проявления, намеченный план обследования был направлен на проведение дифференциального диагноза между внутриутробными инфекциями, врожденным лейкозом и различными формами генодерматозов. Проведенное комплексное обследование позволило исключить врожденные инфекции и гемобластозы.
На фоне проводимой терапии (амоксициллин/клавуланат, внутривенное введение глюкозосолевых растворов, инозин, пиридоксин, никотинамид, рибофлавин) была отмечена некоторая положительная динамика. Кожные проявления стали менее выраженными, некоторые узлы в области спины, груди, конечностей купировались, на их месте остались пигментные пятна. Крупные узлы имели тенденцию к уменьшению в размерах. В динамике клинических проявлений не было отмечено инфекционного процесса, лабораторные показатели оставались в пределах референсных значений. Аппетит у девочки был хороший, питание усваивала в полном объеме, не срыгивала, весовая кривая — положительная.
Однако диагноз оставался неясным, несмотря на привлечение к курации ребенка врачей разных специальностей. Учитывая это, была проведена биопсия узла с последующим морфологическим исследованием, которое позволило выявить типичные признаки склеродермии (рис. 2). Принимая во внимание сроки манифестации заболевания и клинико-морфологические особенности, верифицирована ограниченная нодулярная ВС.

Вопросы, связанные с истинной частотой встречаемости ВС, активно обсуждаются в течение последних 10–15 лет. Это обусловлено тем, что клинические симптомы ВС, обнаруженные при рождении, нередко в течение длительного времени ошибочно трактуются как проявления других заболеваний. Неточная диагностика приводит к существенной задержке назначения адекватной терапии и, как следствие, к прогрессированию заболевания и высокому риску развития осложнений [6–11]. Так, по данным M. Mansour et al. [6], которые проводили международное мультицентровое исследование на протяжении 16 лет (2001–2017 гг.) и смогли выявить 25 педиатрических пациентов с ВС, среднее время от рождения детей с клиническими проявлениями ВС до постановки диагноза составило 2,9 года. При этом у 12 из 25 пациентов к этому времени уже имели место внекожные проявления склеродермии (скелетно-мышечные повреждения отмечены у 80% пациентов, неврологические — у 62%). Аналогичные результаты задержки постановки диагноза представлены и при описании единичных наблюдений ВС [7–11]. Учитывая, что в основе склеродермии лежат аутоиммунные механизмы, а также то, что по-прежнему не исключена вероятность стадийности повреждений различных органов и систем при склеродермии (на ранних стадиях — кожа и подлежащие ткани (ограниченная склеродермия), на поздних — внутренние органы (системная склеродермия)), очень важно своевременно установить диагноз и назначить адекватную терапию [1, 12–16].
Заключение
Все вышесказанное определяет необходимость включать ВС в перечень вероятных заболеваний в тех случаях, когда у новорожденного на коже туловища, и/или конечностей, и/или головы имеют место различные по величине округлые участки гиперемии с цианотичным оттенком на узловатых образованиях, возвышающихся над уровнем кожи. Учитывая, что специфические лабораторные тесты на ВС до настоящего времени не разработаны, окончательная верификация заболевания основывается на характерных гистологических признаках при морфологическом исследовании микропрепаратов, полученных после проведения биопсии измененных участков кожи и подлежащих тканей.
Сведения об авторах:
Ахмина Наталия Ивановна — д.м.н., профессор кафедры неонатологии им. проф. В.В. Гаврюшова ФГБОУ ДПО РМАНПО Минздрава России; 123995, Россия, г. Москва, ул. Баррикадная, д. 2/1; ORCID iD 0000-0002-5870-4938.
Уткин Петр Сергеевич — врач-дерматолог ГБУЗ ДИКБ № 6 ДЗМ; 125438, Россия, г. Москва, 3-й Лихачевский пер., д. 2Б; ORCID iD 0000-0001-5934-8480.
Чабаидзе Жужуна Лазаревна — к.м.н., доцент кафедры неонатологии им. проф. В.В. Гаврюшова ФГБОУ ДПО РМАНПО Минздрава России; 123995, Россия, г. Москва, ул. Баррикадная, д. 2/1; ORCID iD 0000-0002-2192-796X.
Дементьев Александр Анатольевич — к.м.н., доцент кафедры неонатологии им. проф. В.В. Гаврюшова ФГБОУ ДПО РМАНПО Минздрава России; 123995, Россия, г. Москва, ул. Баррикадная, д. 2/1; ORCID iD 0000-0002-7640-1172.
Заплатников Андрей Леонидович — д.м.н., профессор, проректор по учебной работе, заведующий кафедрой неонатологии им. проф. В.В. Гаврюшова, профессор кафедры педиатрии им. акад. Г.Н. Сперанского ФГБОУ ДПО РМАНПО Минздрава России; 123995, Россия, г. Москва, ул. Баррикадная, д. 2/1; ORCID iD 0000-0003-1303-8318.
Прозрачность финансовой деятельности: никто из авторов не имеет финансовой заинтересованности в представленных материалах или методах.
Конфликт интересов отсутствует.
Статья поступила 09.03.2022.
Поступила после рецензирования 01.04.2022.
Принята в печать 26.04.2022.
About the authors:
Nataliya I. Akhmina — Dr. Sc. (Med.), professor of the Prof. V.V. Gavryushov Department of Neonatology, Russian Medical Academy of Continuous Professional Education; 2/1, Barrikadnaya str., Moscow, 125993, Russian Federation; ORCID iD 0000-0002-5870-4938.
Petr S. Utkin — dermatologist, Children’s Infectious Clinical Hospital No. 6; 2B, 3rd Likhachevskiy lane, Moscow, 125438, Russian Federation; ORCID iD 0000-0001-5934-8480.
Zhuzhuna L. Chabaidze — C. Sc. (Med.), associate professor of the Prof. V.V. Gavryushov Department of Neonatology, Russian Medical Academy of Continuous Professional Education; 2/1, Barrikadnaya str., Moscow, 125993, Russian Federation; ORCID iD 0000-0002-2192-796X.
Aleksandr A. Dement’ev — C. Sc. (Med.), associate professor of the Prof. V.V. Gavryushov Department of Neonatology, Russian Medical Academy of Continuous Professional Education; 2/1, Barrikadnaya str., Moscow, 125993, Russian Federation; ORCID iD 0000-0002-7640-1172.
Andrey L. Zaplatnikov — Dr. Sc. (Med.), Professor, Vice-chancellor for Instructional Work, Head of the Prof. N.N. Gavryushov Department of Neonatology, professor of the Acad. G.N. Speranskiy Department of Pediatrics, Russian Medical Academy of Continuous Professional Education; 2/1 Barrikadnaya str., Moscow, 125993, Russian Federation; ORCID iD 0000-0003-1303-8318.
There is no conflict of interests.
Контент доступен под лицензией Creative Commons «Attribution» («Атрибуция») 4.0 Всемирная.
Склеродермия у ребенка
Хотя склеродермия встречается редко и составляет только 5% сосудистых коллагено-зов в детском возрасте, самый распространенный вариант, локализованная склеродермия, или морфеа, может быть едва заметным и, вероятно, не всегда диагностируется.
Локализованная склеродермия может иметь бляшечную, линейную, каплевидную или генерализованную форму. Склеротические бляшки с кремово-белым центром и прогрессирующей границей сиреневого цвета - характерный признак заболевания. Может также развиться выраженная гиперпигментация. В случае чаще всего наблюдаемой формы морфеа на туловище появляется один или несколько очагов диаметром от 1 до 10 см. При генерализованной морфеа подобные очаги распространяются на туловище и конечностях.
Иногда множественные, мелкие, овальные очаги, напоминающие склероатрофический лишай, высыпают на верхней части туловища при каплевидном варианте заболевания. Морфеа может прогрессировать также в линейном варианте, особенно на волосистой части головы, туловище и конечностях. Термином «удар саблей» описывают случаи линейной морфеа, когда от волосистой части головы через лоб и вниз на лицо распространяется вертикально идущая борозда.
При поражении подлежащих мягких тканей и костей может развиться тяжелая деформация. Поражение ЦНС в сочетании с очагом на лице и волосистой части головы, вероятно, встречается чаще, чем ранее предполагалось. Внутричерепные очаги могут привести к тяжелому заболеванию, их необходимо исследовать с помощью МРТ головного мозга. Прогрессирующая гемиатрофия лица (синдром Перри-Ромберга) представляет собой, вероятно, вариант «удара саблей» с минимальным фиброзом, но выраженной атрофией и высоким риском поражения глаз, полости рта и ЦНС. Очаги заживают в течение месяцев и лет с размягчением пораженных участков и атрофией. У детей кожное заболевание без системных симптомов не ассоциируется с прогрессированием в системный склероз.
Лабораторные показатели при локализованной склеродермии неспецифические и прогностическими не являются. Иногда в формуле крови отмечается эозинофилия и может быть повышен ревматоидный фактор. В 10-15% случаев обнаруживаются AHA. Биопсия кожи, взятая из новых очагов или из прогрессирующей границы сформировавшихся бляшек, показывает лимфоцитарное воспаление между пучками коллагена и вокруг кровеносных сосудов в глубоких слоях дермы, которое достигает подкожной жировой клетчатки. Некоторые участки жировой ткани замещены вновь образовавшимся коллагеном.
Склеродермия:
а - подобная бляшка развилась у этой девочки за ушной раковиной
б - линейная морфеа вызвала фиброз и атрофию мягких тканей на IV пальце правой руки у этой молодой женщины
В старых склеротических бляшках гомогенный коллаген простирается от дермо-эпидермального соединения в подкожную жировую клетчатку. Придаточные структуры будто «окутаны» коллагеном, и воспалительного инфильтрата остается мало, или он вообще отсутствует.
К сожалению, попытки терапии не приносят удовлетворительных результатов. Полезность применения пеницилламина, даже при тяжелом локализованном заболевании, никогда не была доказана. Попытки терапии системными антималярийными препаратами, производными витамина D и антиметаболитами давали различные результаты. Короткие курсы высоких доз КСП (1-2 мг/кг в день) могут помочь остановить быстро прогрессирующую линейную морфеа, когда она угрожает важным структурам. Если имеются очаги в ЦНС, некоторые специалисты рекомендуют курсы системных КСП в течение 2-3 мес. с постепенным снижением дозы, а затем в течение 12-18 мес. назначается метотрексат. Недавно стали применять ПУВА-терапию с некоторым успехом для кожных очагов. Если очаги затрагивают суставы, может потребоваться физиотерапия.
Недавно описанный на фоне почечной недостаточности и почечного диализа нефрогенный системный фиброз, известный также как нефрогенная фиброзирующая дермопатия, может совпадать по клиническим признакам с локализованной склеродермией и другими заболеваниями с локализованным фиброзом кожи, которые обсуждаются в конце этой главы. У пациентов типично развивается прогрессирующее утолщение кожи конечностей, латеральных участков туловища и/или спины, лицо и шея при этом не поражаются. Очаги телесного или светло-красного цвета, болезненные, часто с неправильными «амебными» краями. Возможны тяжелые контрактуры и потеря трудоспособности. В последнее десятилетие описано системное поражение с фиброзом глаз, легких, костей, твердой мозговой оболочки и других органов.
У многих пациентов сообщалось о применении контрастных веществ на основе гадолиния, у предрасположенных к фиброзу пациентов этого следует избегать. Гистологические исследования показывают выраженное увеличение похожих на фибробласты веретенообразных клеток и повышенное отложение коллагена и муцина, как при склеромикседеме. Хотя очаги обычно ограничены кожей, сообщалось о редких случаях с поражением легких.
Прогрессирующий системный склероз (ПСС) редко встречается у детей, клинически заболевание протекает так же, как у взрослых. Феномен Рейно наблюдается у 90% пациентов и может предшествовать началу системного заболевания на годы. Классическими кожными признаками являются натянутость кожи в сочетании с деревянистым уплотнением, отек и изменения пигментации. ПСС может привести к угрожающему жизни заболеванию ЖКТ, мышечно-скелетной системы, сердца, легких и почек. У большинства пациентов с ПСС присутствуют антинуклеарные антитела (AHA) и почти у 50% - AHA антинуклеолярного типа свечения. Ряд других антител также обнаруживается в некоторых случаях ПСС и может иметь особое прогностическое значение.
Склеродермия:
а - множественные фиброзные бляшки с изменением пигментации в центре и эритемой по периферии медленно увеличиваются на спине у девочки-подростка
б - атрофическая фиброзная гипопигментированная бляшка простирается вокруг лодыжки у 10-летней девочки в течение 2 лет.
К счастью, девочка не имела функциональных нарушений Склеродермия:
а - у этого подростка развилась липоатрофия на задней поверхности бедра слева
б - редкий вариант локализованной склеродермии по типу «удара саблей» с линейным поражением, идущим от центральной лобной области волосистой части головы через лоб к переносице.
Склеродермия. Причины появления - этиология
а) Пример из истории болезни. 35-летняя женщина обратилась к врачу по поводу очагов блестящей уплотненной кожи на животе, имеющих вид бляшек. Поскольку в целом пациентка была здорова, появление очагов ее удивило и вызвало опасение относительно их распространения по всему телу. Изменения кожи причиняли некоторый дискомфорт, по не были болезненными. Биопсия подтвердила клиническое подозрение на наличие ограниченной склеродермии. Пациентке были назначены клобетазол и кальципотриол местно, после чего состояние кожи несколько улучшилось. Тест на антинуклеарные антитела был положительный, тем не менее, прогрессирующий системный склероз у пациентки не развился.
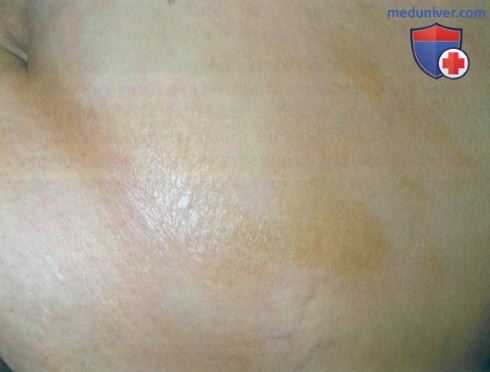
Ограниченная склеродермия на животе у 35-летней женщины
б) Распространенность (эпидемиология):
• Распространенность заболеваний, клинические признаки которых включают склеродермию, составляет 4-253 случая на 1000000 человек.
• Ежегодная заболеваемость системным склерозом в нашей стране составляет 1-2 случая па 100000 человек. Пик начала болезни приходится на 30-50 лет.
• Заболеваемость ограниченной склеродермией (морфеа) оценивается в 25 случаев на 1 млн жителей нашей страны ежегодно.
в) Этиология (причины), патогенез (патология):
• Заболевания, относящиеся к склеродермии, подразделяются на три группы: ограниченная/локализованная склеродермия (морфеа), системный склероз и другие склеродермоподобные заболевания, для которых характерны утолщенные склеротические кожные очаги.
• Самым распространенным сосудистым заболеванием, которое сопутствует системной склеродермии, является болезнь Рейно, проявляющаяся спазмом сосудов пальцев. Характерны изменения цвета от мертвенно-белого до голубоватого (акроцианоз) и, наконец, красного (реперфузионная гиперемия). Феномен Рейно обычно предшествует другим проявлениям болезни, иногда опережая их на несколько лет. У многих пациентов развиваются прогрессирующие структурные изменения кровеносных сосудов малого диаметра, вследствие которых постоянно нарушается кровообращение, что может приводить к изъязвлениям па кончиках пальцев и даже некрозу тканей. Другие сосудистые реакции включают легочную артериальную гипертензию, почечный криз, сосудистую эктазию в антральном отделе желудка.
• Термином «системный склероз» описывается системное заболевание, для которого характерна отечность и уплотнение кожи в сочетании с фиброзом различных тканей и воспалительной инфильтрацией, наблюдающейся во многих внутренних органах. Системный склероз может быть диффузным (ДКСС) или ограниченным кожей и прилежащими к ней тканями (ограниченный кожный системный склероз - ОКСС).
• У пациентов с ограниченным кожными системным склерозом кожи склероз кожи обычно ограничен кистями рук, в меньшей степениоражаются лицо и шея. В некоторых случаях склеродермия со временем развивается в дистальных отделах предплечий. У таких пациентов часто наблюдается CREST-синдром, включающий феномен Рейно, дисфункцию пищевода, склеродактилию, телеангиэктазии и кальциноз кожи.
• У пациентов с диффузный кожный системный склероз на грудной стенке, животе или верхней части плечевого пояса и лопатках часто отмечается склеротическая кожа, которая выглядит как «соль с перцем». У таких пациентов вероятность повреждения внутренних органов вследствие ишемических нарушений или фиброза выше, чем у пациентов с ограниченным кожным системным склерозом или ограниченной склеродермией.
• Почти у 90% пациентов с системным склерозом имеется то, или иное заболевание желудочно-кишечного тракта, однако у половины из этих пациентов симптомы отсутствуют, при этом поражение может затронуть любой отдел желудочно-кишечного тракта. Потенциальные признаки и симптомы включают дисфагию, удушье, изжогу, кашель после глотания, метеоризм, запор и/или диарею, псевдообструкцию, мальабсорбцию и недержание кала. Хронический гастроэзофагеальный рефлюкс и рецидивирующие эпизоды аспирации могут способствовать развитию интерстициальной болезни легких. Сосудистая эктазия в желудке (которую при эндоскопии достаточно часто называют «арбузный желудок») встречается достаточно часто и может привести к гастроинтестинальному кровотечению и анемии.
• Поражениее легких наблюдается более чем у 70% пациентов, обычно оно проявляется диспноэ при нагрузке и непродуктивным кашлем. В нижних сегментах легких при аускультации можно выявить мягкие хрипы по типу «трения замшевой кожи». Заболевания сосудов легких отмечаются у 10-40% пациентов с системным склерозом и наиболее часто встречаются у пациентов с ограниченными кожными поражениями. У пациентов с системной склеродермией пятикратно повышен риск развития рака легких.
• Данные аутопсии подтверждают, что у 60-80% пациентов с диффузными кожным системным склерозом имеется поражение почек. У 50% пациентов наблюдается транзиторная протеинемия, незначительное повышение концентрации креатинина в плазме крови и/или гипертензия. Тяжелое поражение почек развивается у 10-15% пациентов, чаще всего у больных с диффузными кожным системным склерозом.
• Симптоматический перикардит, при котором смертность в течение 5 лег составляет 75%, отмечается у 7-20% пациентов. К основным причинам нарушения сердечной деятельности относятся перикардит, перикардиальный выпот, миокардиальный фиброз, сердечная недостаточность, миокардит, ассоциированный с миозитом, нарушения проводимости и аритмии. Очаговый миокардиальный фиброз является характерным для системного склероза и считается результатом рецидивирующего спазма мелких сосудов. Аритмии являются обычными и в большинстве случаев вызваны фиброзом проводящей системы сердца.
• Заболевания сосудов легких отмечаются у 10-40% пациентов с системной склеродермией и наиболее часто встречаются у пациентов с ограниченным поражением кожи. Оно может наблюдаться при отсутствии выраженного интерстициального воспаления легких, обычно как позднее осложнение, в большинстве случаев прогрессирующее. Могут развиваться тяжелая легочная артериальная гипертензия, иногда с легочной и правожелудочковой сердечной недостаточностью или тромбозом легочных сосудов.
• Отмечается боль, неподвижность и контрактуры суставов, причем чаще всего наблюдаются контрактуры пальцев. Нейропатии и поражение ЦНС могут проявляться головной болью, судорожными припадками, инсультами, поражением сосудов, радикулопатиями и миелопатиями.
• Системная склеродермия вызывает половую дисфункцию у мужчин и женщин. У мужчин она часто приводит к эректильной дисфункции.
Читайте также: