
Аплазия кожи у новорожденного что это
Обновлено: 25.04.2024
Нормальный рост волос и морфология волосяного стержня могут нарушаться при ряде наследственных заболеваний и врожденных синдромов.
Аномалии волос:
I. Алопеция (локализованная).
- Нерубцовая:
Гамартомный невус (например, эпидермальный, сальных желез)
Пигментированный невус
Гемангиомы
«Треугольная» алопеция
- Рубцовая:
Недержание пигмента
Алопеция в «форме венчика» на волосистой части головы
Полосовидная аплазия кожи
II. Гирсутизм (локализованный):
- Врожденный пигментированный невус
- Гладкомышечная гамартома с волосяным компонентом
III. Алопеция (генерализованная):
- Гипогидротическая эктодермальная дисплазия
- Синдром врожденной гемидисплазии с ихтиозиформной эритродермией и дефектами конечностей (CHILD-синдром)
- Синдром Клоустона
- Синдром Коккейна
- Врожденный дискератоз
- Синдром Халлермана-Штрайфа
- Синдром Хэя-Уэллса
- Гомоцистеинурия
- Синдром Менкеса
- Прогерия
- Трихорино-фалангеальный синдром
- Трихотиодистрофия
- Гипоплазия хрящевой ткани и волос
- Синдром Папийона-Лефевра
- Энтеропатический акродерматит
IV. Гирсутизм (генерализованный):
- Синдром липодистрофии Берардинелли
- Синдром цереброокуло-фацио-скелетный
- Синдром Коффина-Сириса
- Гидантоиновый синдром плода
- Фронтометафизеальная дисплазия
- Мукополисахаридозы
- Лепречаунизм
- Синдром Маршалла-Смита
- Трисомия 18
- Синдром шинцеля-Гидиона
- Синдром алкоголизма плода
V. Аномалии стержня волоса:
- Монилетрикс (веретенообразные или четковидные волосы)
- Pili torti (перекрученные волосы):
Синдром Менкеса, болезнь курчавых волос
Синдром шерстистых волос
Невус шерстистых волос
Синдром Базекса
Синдром Крэндалла
Синдром Бьёрнстада
Цинга
- Pili trianguli et canaliculi:
Нерасчесываемые волосы
Эктодермальная дисплазия
- Trichorrhexis invaginata («бамбуковые» волосы):
Синдром Нетертона
VI. Трихотиодистрофия
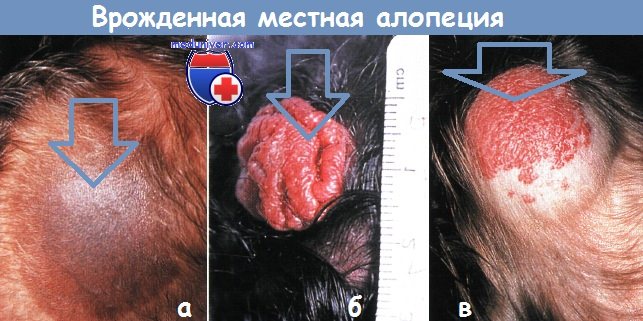
Врожденная локализованная алопеция:
а - выраженное поредение волос наблюдается во врожденном пигментированном невусе на волосистой части головы у 12-месячного ребенка.
Волосы над невусом редкие, темные, длинные и жесткие.
б - участок полной алопеции замечен при рождении в церебриформном невусе сальных желез
в - редкие волосы у годовалого ребенка над регрессирующей гемангиомой в области волосистой части головы
Локализованные пятна алопеции иногда обусловлены перинатальной травмой или гамартомными разрастаниями. Транзиторная потеря волос или перманентное рубцевание в форме кольцевидного венчика вызвано локальным отеком и нарушением сосудов вследствие травмы головы во время родов.
Рубцовая алопеция является частым осложнением врожденной аплазии кожи и недержания пигмента, при которых локализованный спазм сосудов, тромбоз или васкулит приводят к некрозу и изъязвлению кожи. Аплазия кожи может проявляться на волосистой части головы в форме длинных темных волос - признака «волосяного воротничка», который может быть указанием на скрытый дефект невральной трубки.
Ряд невусов, таких как гемангиомы, эпидермальные невусы, пигментированные невусы и невусы соединительной ткани, также могут препятствовать нормальному типу роста волос. Треугольная алопеция, характерная нерубцовая аномалия развития волос может проявиться только в позднем детском или подростковом возрасте. Это состояние проявляется стабильными треугольными, округлыми или прямоугольными залысинами, причем основание очага типично находится в месте соединения лобной и височной зон. Очаги, как правило, односторонние, но иногда встречается билатеральное расположение. Биопсия показывает нормальные веллусные волосы.
Генерализованный скудный или аномальный рост волос указывает на наследственную аномалию стержня волоса или генодерматоз. Монилетрикс - сравнительно частый дефект развития, который приводит к образованию ломких волос, имеющих форму четок. Это состояние наследуется по аутосомно-доминантному типу и клинически проявляется после 2-3-месячного возраста, когда веллусные волосы заменяются аномальными веретенообразными или четковидными волосами. Хотя сильнее всего поражается волосистая часть головы, могут поражаться волосы на любом участке тела. Это состояние персистирует всю жизнь, но в подростковом или взрослом возрасте может наблюдаться улучшение.
Следует быть внимательным, чтобы не перепутать монилетрикс с перекрученными волосами (pili torti), еще одним дефектом, при котором волосы перекручены вокруг своей оси. Этот признак может быть генерализованным или локализованным и проявляется с ростом первых терминальных волос в младенческом возрасте.
Перекрученные волосы могут быть изолированным дефектом волос или наблюдаться в сочетании с мультисистемным поражением, таким как синдром Менкеса (наследственное заболевание обмена меди с поражением ЦНС, сердечно-сосудистой и скелетной систем). Так называемые «бамбуковые волосы» (trichorrhexis invaginata) характеризуются повышенной хрупкостью волосяного стержня и быстрой ломкостью волос, что приводит к генерализованной скудости волосяного покрова. Этот дефект обычно наблюдается при редком аутосомно-рецессивном генодерматозе - синдроме Нетертона, для которого характерны, помимо изменения волос, линейный огибающий ихтиоз и тяжелый атопический дерматит.
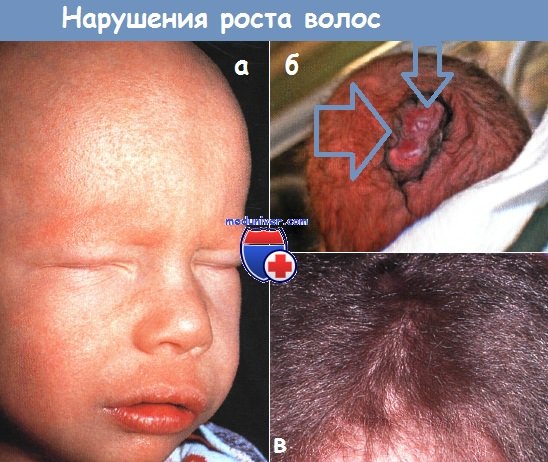
а - надбровная эритема в области бровей и щек. В этом варианте волосяного кератоза воспаление сально-волосяных структур сочетается с прогрессирующей алопецией и атрофией волосяных фолликулов.
Может встречаться как изолированное нарушение кератинизации, так и в сочетании с другими аномалиями.
б - врожденная аплазия кожи с признаком «волосяного воротничка».
У этого недоношенного новорожденного наблюдались две расположенные рядом круглые атрофические фиолетовые бляшки на волосистой части головы с окружающими их густыми волосами.
в - для синдрома нерасчесываемых волос характерны светлые, вьющиеся, непослушные волосы, которые не могут ровно прилегать к плоскости головы, что делает расчесывание таких волос практически невозможным.
Диагноз устанавливается с применением световой микроскопии при обнаружении волос треугольной формы с продольными бороздками вдоль волосяного стержня.
Хотя в некоторых случаях аномалия наследуется по аутосомно-доминантному типу, чаще она возникает спорадически.
При синдромах эктодермальных дисплазий, гетерогенной группы генодерматозов, скудость волосяного покрова сочетается с дизморфиями лица и аномалиями других структур, в том числе ногтей, потовых желез и зубов. При гипогидротических вариантах раннее установление диагноза может спасти жизнь и предотвратить фатальную гипертермию, развивающуюся в ходе детских инфекций, которые в обычных случаях разрешаются самопроизвольно. Трихотиодистрофия - характерный дефект волосяного стержня, который наследуется по аутосомно-рецессивному типу.
Волосы легко расщепляются и отличаются повышенной хрупкостью, низким содержанием серы и чередованием светлой и темной поперечных полос при поляризационной микроскопии. Клинически волосы редкие, тусклые и ломкие. К признакам мультисистемного поражения при трихотиодистрофии относятся внешность по типу прогерии, замедленное психическое развитие, задержка роста, гипогонадизм, фоточувствительность, катаракты и неврологические аномалии.
Детям с врожденными заболеваниями волос необходимы подробное общемедицинское обследование и оценка неврологического статуса. Семейный анамнез помогает установить тип наследования. Волосы исследуются под микроскопом для идентификации специфических аномалий.
Монилетрикс:
а - картина диффузной алопеции с короткими обломанными волосками.
б - при микроскопии наблюдается периодическое сужение волосяных стержней. Волосы хрупкие и обламываются в участках сужения вблизи кожи головы. Trichorrhexis invaginata.
Обратите внимание на напоминающий бамбук внешний вид волосяного стержня у ребенка с синдромом Нетертона. При поляризационной микроскопии на волосах заметны чередующиеся темные и светлые полоски, такие волосы образно называют «тигровыми хвостами». Гипогидротическая эктодермальная дисплазия.
У ребенка отмечаются типичные признаки патологии: редкие волосы, отсутствие бровей и ресниц, лобные бугры, выступающие надбровные дуги, периорбитальная гиперпигментация и толстые вывернутые губы. Синдром Менкеса:
а - у ребенка со светлой кожей, голубыми глазами и редким волосяным покровом отмечаются перекрученные волосы, конвульсии и аномалии развития. В сыворотке выявлены низкие уровни меди и церулоплазмина
б - волосы перекручены вдоль продольной оси.
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
Врожденная аплазия кожи – группа состояний неясной этиологии, которые характеризуются очаговым нарушением формирования кожных покровов с развитием рубцов. Симптомы этого состояния выявляются сразу после рождения ребенка, у которого наблюдается одна или несколько эрозий или изъязвлений на коже головы или, очень редко, на других участках тела. Диагностика врожденной аплазии кожи осуществляется на основании данных осмотра дерматологом, гистологического изучения тканей в очаге поражения. Лечение только симптоматическое с целью недопущения развития вторичной инфекции, но возможна хирургическая коррекция рубцов для уменьшения косметического дефекта.
Общие сведения
Врожденная аплазия кожи – это очаговый дефект развития кожи, при котором нарушается формирование эпидермиса, дермы, придатков, а в особо тяжелых случаях и подкожной клетчатки. Это состояние известно человечеству уже более 250 лет, однако выявить причины его развития до сих пор не удалось, в дерматологии имеются лишь теории на этот счет. Встречаемость врожденной аплазии кожи точно неизвестна, большинство исследователей оценивают ее на уровне 1:10000. Иногда такое состояние сочетается с некоторыми генетическими заболеваниями, другими пороками внутриутробного развития. Врожденная аплазия кожи в большинстве случаев не приводит к тяжелым последствиям, однако косметический дефект в виде рубца на месте патологического очага остается у человека на всю жизнь.
Причины врожденной аплазии кожи
На сегодняшний день нет единой и общепризнанной теории, которая бы объясняла развитие этого врожденного дефекта развития кожи. Предполагается, что причиной врожденной аплазии кожи является целая группа различных патологических факторов, которые ведут к нарушению процесса закрытия нервной трубки или же тормозят развитие эмбриональных зачатков дермы и эпидермиса. Иногда удается выявить семейные формы этого состояния, при этом механизм его наследования, предположительно, аутосомно-доминантный. Но намного чаще встречаются спорадические формы врожденной аплазии кожи, причем иногда в сочетании с другими пороками развития, обусловленные генетическими заболеваниями или воздействием тератогенных факторов. Это дает повод рассматривать данное состояние как следствие влияния на формирующийся плод различных повреждающих факторов.
Симптомы врожденной аплазии кожи
Аплазия кожи выявляется сразу после рождения ребенка. Чаще всего на теменной области обнаруживается очаг округлой формы диаметром 1-3 сантиметра. Примерно в трети случаев возникает два очага, еще реже встречаются три и более участка аплазии кожи. Патологический участок представляет собой эрозию или язву, покрытую корочкой и грануляциями, волосяной покров на нем отсутствует. Однако вокруг язвы вырастают более длинные и темные волоски, что получило название симптома «волосяного воротничка». Цвет образования варьируется от розового до ярко-красного.
Со временем, при отсутствии осложнений (вторичной инфекции, например), участок врожденной аплазии кожи начинает разрешаться с образованием рубца белого цвета. На нем в дальнейшем также не вырастают волосы, и он остается у человека на всю жизнь. Помимо кожных симптомов, у ребенка с очагом врожденной аплазии кожи могут регистрироваться нарушения формирования более глубоко расположенных тканей и другие пороки развития – заячья губа, волчья пасть, атрофия глаз. У детей старшего возраста и взрослых на месте рубца иногда могут развиваться злокачественные новообразования.
Диагностика врожденной аплазии кожи
Распознавание этого заболевания обычно не представляет труда для врача-дерматолога – его симптоматика достаточно специфична, и спутать его с другими врожденными кожными состояниями довольно сложно. Однако в некоторых случаях похожую на врожденную аплазию кожи картину могут иметь и иные патологические процессы и состояния. Поэтому необходимо производить дифференциальную диагностику этой патологии с такими заболеваниями, как очаговая склеродермия, дискоидная красная волчанка, а также с последствиями перинатальной травмы (от щипцов и других акушерских инструментов). Очень похожи на аплазию семейные формы гипоплазии кожи лица, однако при этом атрофические очаги наблюдаются в области висков.
Наиболее точные диагностические данные может дать гистологическое изучение тканей патологического очага. При врожденной аплазии кожи наблюдается резкое уменьшение толщины (вплоть до 1-го слоя клеток) эпидермиса, дермы, иногда подкожной клетчатки. Признаков воспаления и лейкоцитарной инфильтрации (при отсутствии вторичной инфекции) не наблюдается, также не выявляют придатков кожи.
Лечение и прогноз врожденной аплазии кожи
Лечение врожденной аплазии кожи условно разделяется на два этапа. Первый производится непосредственно после рождения ребенка – в этот период показаны только профилактические и уходовые мероприятия (обработка эрозий антибактериальными мазями, увлажняющими средствами), наложение повязки для уменьшения риска травмирования. Через несколько недель на месте патологического очага сформируется рубец, который хоть и остается на всю жизнь, но может быть прикрыт окружающими волосами. Второй этап сводится к хирургическому устранению дефекта (чаще всего, по косметическим причинам), и проводить его можно в позднем детском или взрослом возрасте. При коррекции значительных по площади участков врожденной аплазии кожи может применяться пересадка кожи. Прогноз заболевания в целом благоприятный, некоторые исследователи указывают на необходимость ежегодного осмотра рубца у дерматолога из-за риска развития онкологических процессов.
Агенезия мозолистого тела — это врожденное отсутствие мозолистого тела либо его части. Аномалия обусловлена генетическими нарушениями, сосудистыми мальформациями, тератогенными факторами. Основные признаки заболевания: двигательные расстройства, задержка психоречевого развития, судорожные приступы. При негрубом (частичном) варианте патологии возможно малосимптомное течение. Для диагностики состояния назначается церебральные КТ или МРТ, нейросонография у новорожденных, генетические исследования. Лечение симптоматическое: медикаментозная коррекция осложнений, реабилитационные программы.
МКБ-10
Общие сведения
Агенезия мозолистого тела (АМТ) — один из наиболее частых пороков нервной системы. Распространенность болезни в популяции составляет от 0,05% до 7% среди новорожденных, причем в группе детей с замедленным становлением психики агенезия встречается у 2,3%. Калифорнийская программа по изучению врожденных пороков предоставляет другие данные по частоте агенезии — 1,4 на 10000 живых новорожденных. Впервые состояние было описано в 1812 году в ходе аутопсии, проведенной немецким анатомом И. Рэйлем, и названо «природной моделью рассеченного мозга».
Причины
Точные этиологические факторы заболевания не установлены. В современной неврологии преобладает мультифакториальная теория, согласно которой для формирования врожденного порока ЦНС требуется комбинация неблагоприятных экзогенных и эндогенных причин. Ученые выделяют несколько наиболее вероятных предпосылок развития агенезии:
- Генетические аномалии. Повреждения мозолистого тела отмечаются при различных наследственных синдромах: Миллер-Дикера, Рубинштейна-Тауби, Доннаи-Кугана. Состояние входит в состав не менее 7 аутосомно-доминантных, 23 аутосомно-рецессивных, 12 Х-сцепленных врожденных заболеваний.
- Сосудистые нарушения. Причиной недоразвития мозолистого тела могут выступать артериовенозные мальформации или аневризмы, которые характеризуются отсутствием нормальной капиллярной сети. При этом возникает феномен обкрадывания, клетки МТ не получают должного количества кислорода, питательных веществ.
- Токсические влияния. Болезнь связана с действием тератогенных химических факторов: лекарственных препаратов, солей тяжелых металлов, пестицидов и бытовой химии. Негативное влияние на формирование ЦНС плода оказывает вдыхание табачного дыма (активное или пассивное курение) или прием беременной алкоголя во время гестации.
- Внутриутробные инфекции. Аномалии формирования неврологических структур, в том числе агенезия мозолистого тела, встречаются при проникновении возбудителей в организм плода на 2-3 месяце беременности. Нейротропные свойства демонстрируют герпетические инфекции, токсоплазмоз, цитомегаловирус.
Основным фактором риска выступает недоношенность. У новорожденных, родившихся до 27-недельного срока гестации МТ истончено в задних отделах, между 28 и 30 неделями — только в области валика. У рожденных после 30 недели в неонатальном периоде изменения не обнаруживаются, хотя при нейропсихологическом исследовании у школьников зачастую выявляется дефицит межполушарной передачи познавательной информации.
Патогенез
Агенезия возникает при нарушении дифференциации нервной трубки в период со 2 до 5 месяца внутриутробного развития. При полном отсутствии МТ третий мозговой желудочек остается открытым, не формируются столбы свода мозга, отсутствуют прозрачные перегородки. В 60% случаев при АМТ передней комиссуры нет вообще. В 10% она увеличена и берет на себя часть функций мозолистого тела у новорожденных, а также на следующих этапах постнатального периода.
Характерным анатомическим изменением является колпоцефалия, при которой расширены задние отделы боковых церебральных желудочков. Состояние не относится к истинной гидроцефалии новорожденных, а обусловлено уменьшением кортикальных ассоциативных путей. Еще один типичный признак порока — пучки Пробста, представляющие собой неправильно ориентированные аксоны, расположенные параллельно межполушарной щели.
Классификация
В практической неврологии состояние подразделяют на тотальное, когда орган полностью отсутствует, и частичное (парциальное), при котором визуализационные методы не обнаруживают отдельные участки МТ. Это имеет решающее значение для тяжести клинической картины, возможных осложнений. В соответствии с патогенетическими особенностями формирования врожденных пороков, выделяют следующие 3 формы болезни:
- Агенезия. Закладка эмбрионального зачатка МТ отсутствует полностью.
- Аплазия. Эмбриональный зачаток мозолистого тела есть, но не развивается.
- Гипоплазия. МТ недостаточно развито из-за нарушений на одном из этапов эмбриогенеза: размеры и масса органа уменьшены, его функциональная активность снижена.
Симптомы
Клиническая картина агенезии мозолистого тела широко варьирует от практически бессимптомных форм (при гипоплазии) до критических нервно-психических расстройств при его грубом недоразвитии, сопровождающемся другими врожденными пороками ЦНС. У новорожденных признаки патологии могут вовсе отсутствовать и проявляться по мере взросления младенца задержкой психомоторного развития.
Двигательные нарушения определяются у 35-40% пациентов. Они проявляются мышечной гипотонией или дистонией, гипер- или гипорефлексией, нарушением глотательного и сосательного рефлексов. Дети позже начинают держать голову, испытывают затруднения при обучении сидению, ползанию, ходьбе. Могут отмечаться координационные нарушения, неуклюжая походка. Из пароксизмальных расстройств у новорожденных и детей первого года жизни преобладают судороги.
Мозолистое тело поддерживает связь между церебральными зонами, формирует межполушарную организацию высших психических процессов. При его агенезии либо гипоплазии у детей выявляются когнитивные расстройства. У новорожденных пациентов и в раннем детстве наблюдается задержка речи, снижение динамического компонента игровой деятельности. В дошкольном и школьном возрасте возникают проблемы с концентрацией внимания, расстройства памяти, при тотальной АМТ снижен коэффициент интеллекта.
Осложнения
Около 65% случаев заболевания сопровождаются сопутствующими врожденными патологиями, среди которых преобладают мальформации кортикального развития (22,8%), межполушарные кисты (14,3%), голопрозэнцефалия (14,3%). К более редким сопутствующим аномалиям относят кисты и гипоплазию мозжечка, синдром Арнольда-Киари. До 20% новорожденных, кроме структур ЦНС, имеют пороки нескольких внутренних органов.
У 75% больных с тотальным поражением наблюдается симптоматическая эпилепсия височно-лобной локализации, в 66% случаев выражены когнитивные нарушения. У 16% пациентов формируются расстройства аутистического спектра. Изредка встречаются патологии органа зрения в виде хориоретинальных лакунарных очагов, сочетанной аномалии зрительных нервов.
Диагностика
В качестве первичного метода обследования в пренатальном периоде проводится акушерское УЗИ. У новорожденных для скрининговой диагностики используется нейросонография, однако этот метод не всегда показывает хорошую информативность, особенно при парциальной агенезии. Для верификации диагноза назначаются следующие методы исследования:
- КТ головного мозга. При компьютерной томографии определяются широко расставленные передние рога, высокое стояние третьего желудочка, параллельный ход медиальных стенок боковых желудочков. КТ производится в рамках постнатальной диагностики.
- МРТ головного мозга. Для максимально точной визуализации степени агенезии или гипоплазии мозолистого тела новорожденным выполняется магнитно-резонансная томография в трех плоскостях. По показаниям МРТ может рекомендоваться беременным женщинам для исключения несовместимых с жизнью сочетанных пороков ЦНС.
- Нейропсихологическое обследование. Для изучения когнитивных функций у детей применяется шкала интеллекта Векслера (WISC-Revised), адаптированное чтение и правописание (Schonnel Graded Reading and Spelling Tests), оценка вербальной беглости, тест контролируемых устных ассоциаций (Controlled Oral Word Association Test).
- Генетический анализ. Для подтверждения или исключения наследственных заболеваний, сопровождающихся агенезией мозолистого тела, показаны кариотипирование, секвенирование генома, проводимое как новорожденным, так и детям другого возраста. Исследования также проводят в антенатальном периоде для принятия решения о сохранении или прерывании беременности.
Лечение агенезии мозолистого тела
Специфическая терапия отсутствует. Медикаментозное лечение назначается неонатологом или педиатром индивидуально с учетом ведущих патологических синдромов: у новорожденных, детей раннего возраста используются антиконвульсанты, нейрометаболические препараты, дегидратационная терапия. Основу медицинской помощи составляет комплексная реабилитация, которая включает следующие составляющие:
- Нейрологопедические программы. Занятия с детским логопедом проводятся для становления речевой функции, ликвидации проявлений дизартрии, улучшения артикуляции.
- Дефектологические программы. Помощь коррекционных педагогов требуется детям с интеллектуальными нарушениями, которые не могут проходить обучение в обычных классах.
- Нейроакустические программы. Формирование и гармонизация высших психических функций производятся с помощью звуковой терапии, музыкотерапии.
Прогноз и профилактика
Прогноз определяется видом врожденной аномалии мозолистого тела, наличием сопутствующих пороков развития ЦНС. Благоприятный исход наблюдается при частичной гипоплазии МТ, а в случае комбинированных церебральных пороков у новорожденных могут быть жизнеугрожающие осложнения. Профилактические меры включают медико-генетическое консультирование, исключение тератогенных влияний в гестационном периоде.
1. Эпилептические проявления когнитивные и аутистические расстройства у пациентов с агенезией мозолистого тела: результаты нейропсихологического тестирования/ О.А. Милованова, О.А. Комиссарова, Т.Ю. Тараканова, С.В. Бугрий// Эпилепсия и пароксизмальные состояния. — 2018. — №4.
2. Влияние особенностей строения мозолистого тела и доминирующего полушария на протекание психических процессов в юношеском возрасте/ У.С. Чернышова, Т.Ю. Хабарова, Д.А. Соколов// Центральный научный вестник. — 2016.
4. Молекулярная эмбриология: на пути каталогизации генов врожденных пороков развития головного мозга/ В.П. Пишак, М.А. Ризничук// Международный журнал педиатрии, акушерства и гинекологии. — 2014. — №5.
Врожденная аплазия кожи у новорожденного
Врожденная аплазия кожи - гетерогенная группа заболеваний, общим признаком которых является врожденное отсутствие кожи. Хотя причина аплазии неизвестна, само состояние регистрируется вот уже более 250 лет.
В классической форме аплазия кожи проявляется одной или несколькими эрозиями или изъязвлениями, покрытыми коркой или тонкой оболочкой, на макушке волосистой части головы. Заживление в форме атрофических, лишенных волос рубцов происходит в течение 2-6 мес. в зависимости от размера и глубины изъязвлений (которые могут распространяться на всю глубину мягких тканей вплоть до кости).
Это доброкачественное состояние может наследоваться по аутосомно-доминантному признаку или возникать спорадически. Реже встречается поражение туловища и конечностей, такие очаги могут ассоциироваться с дефектами конечностей, БЭ и хромосомными аномалиями. Очаг на волосистой части головы с кольцом окружающих его длинных темных волос (признак «волосяного воротничка») представляет собой дефект вследствие неполного заращения невральной трубки. Для оценки возможной связи с подлежащими участками мозга и кости показана МРТ.
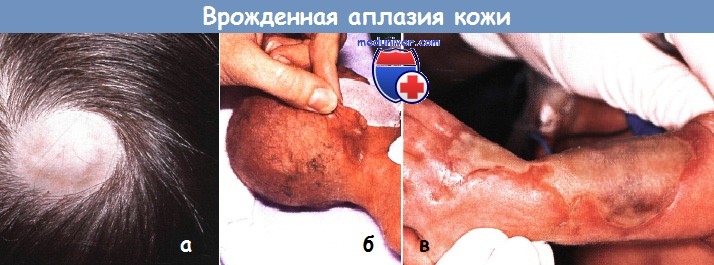
Врожденная аплазия кожи:
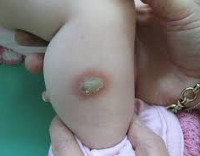
а - изолированный дефект кожи наблюдался на руке новорожденного
б - у этого недоношенного ребенка с небольшим дефектом на задней части шеи гемиатрофия правой стороны тела (синдром Адамса-Оливера).
При неосложненной врожденной аплазии кожи дефект корректируется простым иссечением в позднем детском или во взрослом возрасте. В случае крупных очагов может потребоваться поэтапное иссечение и применение тканевых экспандеров. У новорожденных необходимо оценить глубину дефекта, предупредить повреждение ткани и инфекцию и обследовать ребенка на наличие сочетанных аномалий.
Мягкие компрессы с физиологическим раствором, местное применение антибиотиков и наложение стерильных повязок являются адекватными терапевтическими мероприятиями для большинства пациентов. Для ведения пациентов с крупными дефектами применяются герметичные повязки (Duoderm®, Comfeel®). Иногда необходимо раннее хирургическое вмешательство.
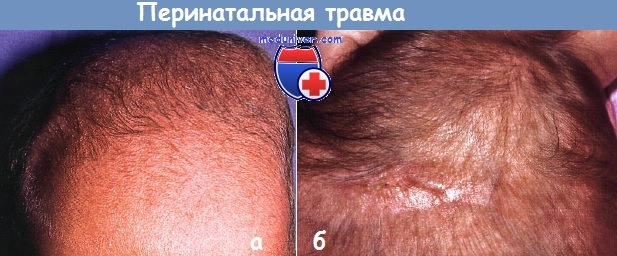
Перинатальная травма:
а - цефалогематома. Просачивание крови под надкостницу, обычно разрешается без особенностей.
б - редких случаях перинатальная травма приводит к образованию кольцевидного венчика на волосистой части головы с рубцеванием и перманентной утратой волос.
Перинатальная травма головы может привести к подобным дефектам, которые обнаруживают при рождении или в первый месяц жизни. Взятие крови в области волосистой части головы, электроды монитора и щипцы могут привести к образованию небольших язвочек, которые заживают с рубцеванием.
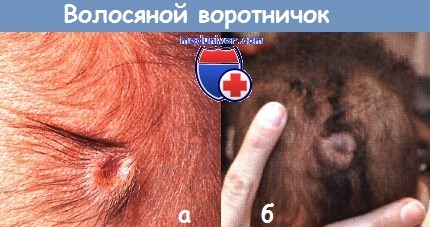
а - признак «волосяного воротничка» на волосистой части головы у новорожденного.
б - у этого здорового 4-месячного ребенка признак «волосяного воротничка».
МРТ не вывила никакой аномалии подлежащих структур.
Безволосый кольцевидный венчик на волосистой части головы - еще одна форма локализованной травмы головы, в редких случаях ассоциирующейся с родовой опухолью и цефалгематомой, которая иногда разрешается с рубцовой алопецией.
Врожденную аплазию кожи напоминают другие невоидные дефекты, часто проявляющиеся в форме лишенных волос пятен на волосистой части головы. Однако эти родимые пятна не покрываются коркой и не бывают атрофическими и обычно имеют характерные клинические признаки. Некоторые врожденные, слабо пигментированные пигментные невусы на волосистой части головы также могут имитировать врожденную аплазию кожи.
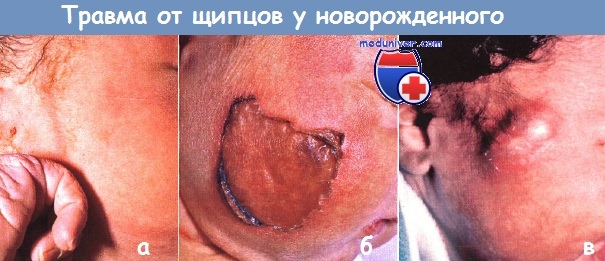
Дефекты, похожие на врожденную аплазию кожи:
а - травмы от щипцов чаще всего ассоциируются с образованием гематомы без нарушения кожных покровов.
Однако в редких случаях случается (б) поверхностный некроз и (в) жировой некроз.
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
Дефект кожи головы - методы диагностики, лечения по Европейским рекомендациям
Синдром Адамса-Оливера включает врожденную аплазию кожи (ВАК) черепа с терминальным поперечным поражением конечностей с дефектом конечностей. Один из четырех случаев трисомии 13-15 проявляется врожденной аплазией кожи (ВАК).
Распространенность врожденной аплазии кожи (ВАК) в настоящее время неизвестна, хотя она была оценена в 3 случая на 10000 рождений. В 60% случаев, дефект ограничен волосистой частью головы, в то время как оставшиеся 40% могут быть рассеяны по телу или конечностям. Комбинированные поражения волосистой части головы и остальных частей тела обнаружены у 25% пациентов. Около 80% кожи головы занимают дефекты срединной линии, в основном на вершине черепа, хотя они могут занимать и боковое положение. Приблизительно 20% случаев ВАК головы связаны с подлежащим дефектом черепа.
Происхождение врожденной аплазии кожи (ВАК) головы остается неизвестным:
1. Некоторые авторы рассматривают это состояние как часть ДНТ и предполагают, что оно представляет форму дефекта закрытия нервной трубки, похожей на грыжу и менингоцеле.
2. Другие приписывают врожденную аплазию кожи (ВАК) головы нарушению слияния мезодермы, которое необходимо для нормального развития эктодермы.
3. Врожденная аплазия кожи (ВАК) также были отнесены к дефекту кровотока на вершине черепа (критическая зона для васкуляризации кожи головы) из-за скудного развития сосудов или к нарушению кровотока при перерастяжении кожи головы плода.
4. Врожденная аплазия кожи (ВАК) связывают и с ишемическими или тромботическими эпизодами в плаценте.
5. Другие относят эти дефекты к очаговым некрозам от давления как это происходит при амниотическом синдроме при клинически узком тазе.
6. Врожденная аплазия кожи (ВАК) и дефекты черепа также упоминаются в ассоциации с тератогенами (мизопростол, метимазол, аминоптерин, метотрексат и бензодиазепины), внутриутробными инфекциями (ветряная оспа или вирус простого герпеса) и злоупотреблением кокаином.
Диагностика врожденной аплазии кожи (ВАК) кожи головы выполняется при осмотре, мы рекомендуем использовать хорошее освещение и увеличительные лупы. Макроскопически поражения хорошо ограничены, имеют овальную или неправильную форму с размером 0,5-10 см по типу язвы неравной глубины. Когда повреждения заживают до рождения, они выглядят как врожденные шрамы. Микроскопически поражения состоят из атрофических кожных элементов без кожных придатков. Тщательное обследование новорожденных обязательно для исключения других кожных или системных аномалий.
Случаи с несколькими пороками развития предоставляются для соответствующих генетических исследований, такие состояния при нейрохирургической консультации, обычно требуют нейровизуализации. МРТ позволяет оценить состояние головного мозга. МР-ангиография и флебография используются для описания состояния магистральных сосудов и сагиттального синуса. Для оценки дефектов черепа используется КТ с трехмерной реконструкцией.
Дифференциальный диагноз устанавливается между атретической цефалоцеле, sinus pericranii и гетеротопической тканью. Некоторые поражениях кожи головы могут быть ошибочно отнесены к травматическим после вакуум-экстракции, применения щипцов или мониторинга электродами кожи головы плода.
Два основных варианта лечения врожденной аплазии кожи (ВАК) кожи головы:
1. Консервативное лечение. Маленький боковой или поверхностный дефект кожи (не связанный с черепом или твердой мозговой оболочкой) ведется консервативно в течение нескольких недель. Антибиотики используются только при наличии кожной инфекции.
2. Хирургическое лечение. Начало лечения зависит от следующих факторов:
а) локализация: дефекты срединной линии более склонны к развитию кровоизлияния или тромбоза сагиттального синуса.
б) размер поражения: мелкие дефекты кожи проще лечатся прямым закрытием или перемещением кожных лоскутов. Этот подход является предпочтительным, обеспечивая нормальный вид волосяного покрова.
в) глубина поражения: ВАК может поражать надкостницу, череп и твердую мозговую оболочку. Открытый мозг более подвержен менингиту, энцефалиту и кровотечению из сагиттального синуса. Большие дефекты требуют более сложных кожных лоскутов и даже пересадки кожи.
г) общее состояние и продолжительность жизни детей также должны быть приняты во внимание.
Время проведения хирургической коррекции определяется с учетом всех факторов. К участию в операции необходимо привлечь пластического хирурга. Прогноз при ВАК головы, как правило, хороший у большинства пациентов, и зависит от размеров поражения и сопутствующих аномалий, однако при больших и сложных поражениях смертность может превышать 50%.
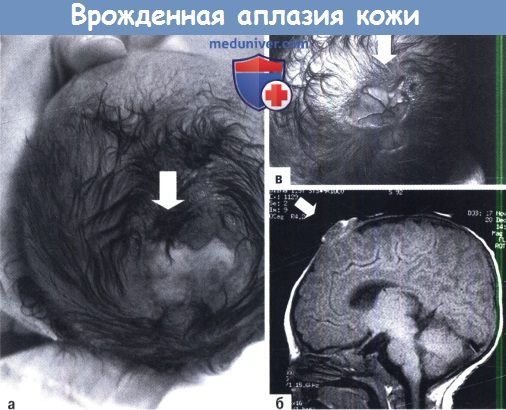
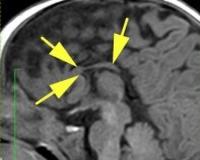
А, Б. Два случая врожденной аплазии кожи черепа (белая стрелка).
В. МРТ незаращенного повреждения врожденной аплазии, зарождение грыжи мозга (перепечатано с разрешения из Child’s Nervous System, Springer-Verlag).
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
Читайте также: